蛋白质聚集的分子动力学模拟
( Molecular dynamics simulations of protein aggregation)
1.MD 模拟和基本分析
以 Aβ16-22 为例,其两端都带有封端基团,序列为 ACE-KLVFFAE-NME。由于蛋白质数据库 (PDB) 不包含该肽的结构,因此可以从 Aβ42 的 PDB 结构的残基 16-21 的坐标中检索以下模拟的起始结构,例如 PDB ID 1Z0Q。在 PyMOL 的蛋白质模式下使用 Builder 工具,ACE 和 NME 封端组可以分别添加到 N 和 C 端。

1.1 六肽模拟体系建立
1. 第一步是为 Aβ16-22 单体产生一个松弛的构象。对于 Aβ16-22,建议模拟长度为 1 μs 或更长,使用构象聚类确定最稳定的单体结构(本文省略这步)。
packmol < packmol.inp
或者使用gmx insert-molecules命令
gmx insert-molecules -ci abeta16-22.pdb -nmol 6 -box 10 10 10 -o abeta16-22_hexamer.pdb
1.2 不同的模拟步骤创建目录
mkdir 1-topol 2-em 3-nvt 4-npt 5-md mdp
1.3 拓扑构建
拓扑文件包含有关将被模拟的分子类型和分子数量的信息。上一步的 .pdb 文件作为输入,除了拓扑文件之外,还会生成一个 .gro 文件,该文件与 .pdb 文件一样,也包含模拟系统的坐标。它们之间的主要区别在于它们的格式。
cd 1-topol/
(2)运行GROMACS pdb2gmx 命令处理输入的结构文件并创建扩展名为.top 的拓扑文件、扩展名为.itp 的拓扑包含文件和扩展名为.itp 的位置约束文件。
gmx pdb2gmx -f ../abeta16-22_hexamer.pdb -o protein.gro -p topol.top -ignh -ter <<EOF
1
1
3
4
3
4
3
4
3
4
3
4
3
4
EOF
选项说明:
-f: 读取输入结构文件 abeta16-22_hexamer.pdb
-o and -p: 写入输出结构文件 protein.gro 和系统拓扑文件 topol.top
-ignh: 忽略输入文件中的氢原子,这是可取的,因为输入文件和力场中氢原子的命名约定不同。GROMACS 将使用所选力场的 H 原子名称添加新的氢原子。
-ter: N 端和 C 端的质子化状态可以-ter 标志交互选择
Option 1: choosing protein force field (charmm36-mar2019)
Option 1: choosing water force field (TIP3P)
Option 3: choosing N-terminus (None, as we use ACE capping)
Option 4: choosing C-terminus (None, as we use NME capping)
Options 3 and 4 are repeated for each peptide in the system, in this example six.
(3) 接下来,创建一个模拟框。请注意,上面定义的框仅用于放置肽。和以前一样,选择了一个 10 × 10 × 10 nm3 的立方体盒子:
gmx editconf -f protein.gro -o box.gro -bt cubic -box 10 10 10
(4)在模拟盒里装入水分子
gmx grompp -f ../mdp/ions.mdp -c protein-solvated.gro -p topol.top -o protein-ions.tpr
echo 13 | gmx genion -s protein-ions.tpr -o protein-ions.gro -p topol -neutral -conc 0.15

图 1 六个 Aβ16-22 肽(显示为不同颜色),随机放置在水盒子中
上面我们准备了模拟的拓扑文件和坐标文件,接下来运行MD模拟。
1. 能量最小化
(1)对于能量最小化 (EM) 步骤,切换到目录 2-em:
cd ../2-em
(2)如 em.mdp 文件所示,我们采用最速下降法来最小化系统,直到最大力为达到 100 kJ/mol/nm 或 2,000 个最小化步骤。grompp 用于将结构、拓扑和仿真参数组合成二进制输入文件 protein-em.tpr,然后将其传递给 GROMACS mdrun 命令。
gmx grompp -f ../mdp/em.mdp -c ../1-topol/protein-ions.gro -p ../1-topol/topol.top -o protein-em.tpr
gmx mdrun -v -deffnm protein-em
成功执行 mdrun 命令后,会生成以下文件:
2. NVT 平衡
cd ../3-nvt
gmx grompp -f ../mdp/nvt.mdp -c ../1-topol/protein-em.gro -p ../1-topol/topol.top -o protein-nvt.tpr -r ../2-em/protein-em.gro
gmx mdrun -v -deffnm protein-nvt
3. NPT 平衡
cd ../4-npt
gmx grompp -f ../mdp/npt.mdp -c ../1-topol/protein-nvt.gro -p ../1-topol/topol.top -o protein-npt.tpr -r ../3-nvt/protein-nvt.gro
gmx mdrun -v -deffnm protein-npt
4. MD 生产运行
要执行本例中的MD 生产模拟,切换到相应目录
cd ../5-md
gmx grompp -f ../mdp/md.mdp -c ../1-topol/protein-npt.gro -p ../1-topol/topol.top -o protein-md.tpr -r ../4-npt/protein-npt.gro
gmx mdrun -v -deffnm protein-md
1. Anaconda创建环境
conda create -n conda-python3.6 python=3.6
conda activate conda-python3.6
安装MDanalysis 和MDtraj
conda install -c conda-forge mdanalysis
conda install -c omnia mdtraj
2. 为分析创建一个新目录
cd ../
mkdir analysis
cd analysis/
MD轨迹处理
(1)将轨迹文件 protein_md.xtc 和运行输入文件 protein_md.tpr 从生产 MD 目录复制到分析目录。
cp ../5-md/ protein_md.xtc ./
cp ../5-md/ protein_md.tpr ./
(2)对于分析,我们只需要蛋白质的坐标,而不需要溶剂和离子的坐标。使用 GROMACS trjconv 命令执行提取和重新保存:
(3)我们还需要以 .gro 或 .pdb 格式提取参考结构文件。在当前示例中,我们创建了一个 .pdb 文件。
gmx trjconv -s protein_md.tpr -f protein_md.xtc -o protein_only.pdb -dump 0
On the command prompt: select
option “1” for output “protein”
Explanation: -dump 0: 储轨迹文件的第一帧。
(4)需要在分析之前解决的蛋白质聚集模拟的一个特殊问题是 MD 模拟过程中的 PBC,它可能导致蛋白质似乎被破坏。许多分析脚本无法处理此类破碎的蛋白质,这会导致分析中出现假象。在 VMD 中读入新创建protein_only.pdb 和 protein_only.xtc 文件并可视化轨迹。然后在“Extensions”选项卡上打开 Tk 控制台,并通过输入命令可视化蛋白质系统周围的 PBC 框:
pbc box
[atomselect top all] set chain 0
pbc join fragment -all
另存为:protein-nopbc.trr
或者使用trjconv处理PBC
gmx trjconv -s protein_only.pdb -f protein_only.xtc -pbc nojump -o protein_nopbc.trr
4. 计算低聚状态和残基间接触频率
conda activate conda-python3.6
python oligos-cmap.py protein_only.pdb protein_nopbc.trr 4
python plot-cmap.py protein_only.pdb protein_nopbc.trr contact-map.dat
python plot-oligostate.py protein_only.pdb protein_nopbc.trr oligo-highest-size.dat 100
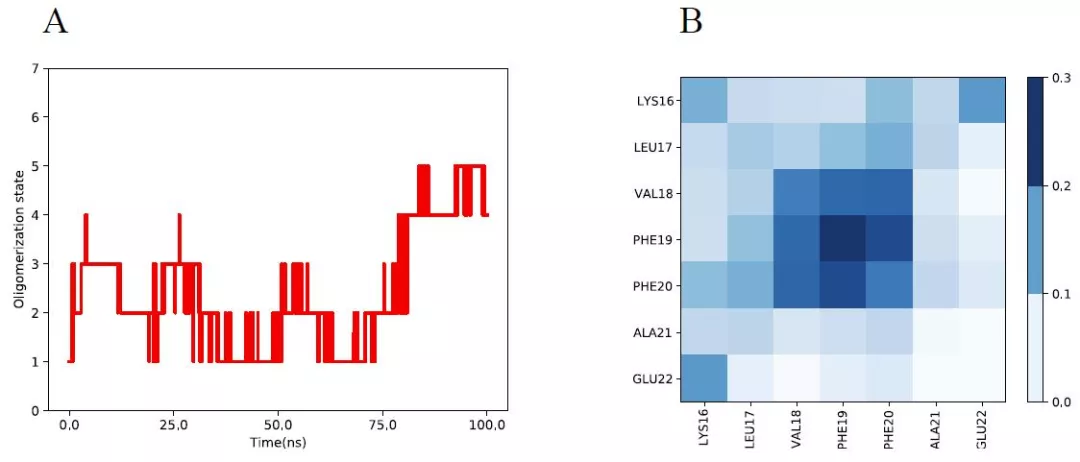
图 2 6 个 Aβ16-22 肽聚集后的 100 ns MD 模拟分析
(A)显示了系统随时间的低聚状态;(B)具有概率的残基间接触图。







order desloratadine 5mg for sale – where can i buy beclomethasone buy albuterol generic
depo-medrol otc – buy fml-forte no prescription astelin 10ml tablet
purchase ventolin without prescription – buy promethazine without prescription theo-24 Cr ca
ivermectina 6mg – buy cefaclor generic cefaclor 250mg pill
amoxil without prescription – erythromycin over the counter purchase ciprofloxacin pill
augmentin order – ampicillin ca ciprofloxacin pill
generic atarax 10mg – pamelor tablet purchase amitriptyline without prescription
order quetiapine 100mg for sale – order ziprasidone 40mg without prescription order eskalith sale
clomipramine online – order citalopram how to buy doxepin
buy zidovudine – zyloprim 100mg oral
clozapine 50mg over the counter – buy generic frumil online pepcid 20mg over the counter
order glycomet 500mg sale – metformin 500mg usa lincomycin tablet
purchase lasix pill – prazosin sale captopril buy online
flagyl 400mg usa – oral cefaclor 250mg brand azithromycin
how to get acillin without a prescription buy acillin without prescription amoxicillin uk
order valacyclovir 500mg generic – buy cheap zovirax zovirax price
ivermectin 6mg stromectol – buy sumycin online cheap buy generic tetracycline 250mg
stromectol generic – where can i buy ciprofloxacin buy sumycin without prescription
order generic ciplox 500 mg – buy ciprofloxacin buy erythromycin sale
brand cipro 1000mg – brand keflex order augmentin 375mg for sale
cipro 500mg tablet – oral keflex 500mg augmentin 1000mg for sale
lipitor 80mg usa lipitor without prescription lipitor 20mg oral