嘉宾:章程博士,伦敦大学学院
主持人:惠成功
时间:2019年9月3日晚20:00
摘要:
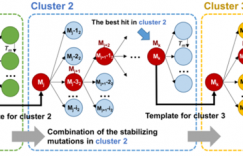
Computationally guided semirational design has significant potential for improving the aggregation kinetics of protein biopharmaceuticals. While improvement in the global conformational stability can stabilize proteins to aggregation under some conditions, previous studies suggest that such an approach is limited, because thermal transition temperatures ( Tm) and the fraction of protein unfolded ( fT) tend to only correlate with aggregation kinetics where the protein is incubated at temperatures approaching the Tm. This is because under these conditions, aggregation from globally unfolded protein becomes dominant. However, under native conditions, the aggregation kinetics are presumed to be dependent on local structural fluctuations or partial unfolding of the native state, which reveal regions of high propensity to form protein-protein interactions that lead to aggregation. In this work, we have targeted the design of stabilizing mutations to regions of the A33 Fab surface structure, which were predicted to be more flexible. This Fab already has high global stability, and global unfolding is not the main cause of aggregation under most conditions. Therefore, the aim was to reduce the conformational flexibility and entropy of the native protein at various locations and thus identify which of those regions has the greatest influence on the aggregation kinetics. Highly dynamic regions of structure were identified through both molecular dynamics simulation and B-factor analysis of related X-ray crystal structures. The most flexible residues were mutated into more stable variants, as predicted by Rosetta, which evaluates the ΔΔGND for each potential point mutation. Additional destabilizing variants were prepared as controls to evaluate the prediction accuracy and also to assess the general influence of conformational stability on aggregation kinetics. The thermal conformational stability, and aggregation rates of 18 variants at 65 °C, were each examined at pH 4, 200 mM ionic strength, under which conditions the initial wild-type protein was <5% unfolded. Variants with decreased Tm values led to more rapid aggregation due to an increase in the fraction of protein unfolded under the conditions studied. As expected, no significant improvements were observed in the global conformational stability as measured by Tm. However, 6 of the 12 stable variants led to an increase in the cooperativity of unfolding, consistent with lower conformational flexibility and entropy in the native ensemble. Three of these had 5-11% lower aggregation rates, and their structural clustering indicated that the local dynamics of the C-terminus of the heavy chain had a role in influencing the aggregation rate.
KEY WORDS:
Fab; aggregation; cooperativity; entropy; global unfolding; melting temperature (Tm); molecular dynamics; mutagenesis; protein engineering; thermal stabilit






order semaglutide online cheap – buy semaglutide medication buy DDAVP
order prandin without prescription – how to get jardiance without a prescription empagliflozin 25mg cost
buy cheap glycomet – sitagliptin 100 mg brand order acarbose 50mg pills
order glyburide online – actos 15mg cost buy forxiga 10mg for sale
Wow, incredible weblog layout! How lengthy have you ever been blogging for?
you made running a blog glance easy. The total glance of your
website is magnificent, let alone the content! You can see similar
here dobry sklep
desloratadine for sale online – buy aristocort pills for sale buy antihistamine pills onlin
depo-medrol without a doctor prescription – buy montelukast online buy azelastine generic
ventolin online – theo-24 Cr online theophylline online order
cleocin 150mg sale – monodox cheap chloramphenicol generic
zithromax price – how to buy ofloxacin oral ciplox
amoxicillin uk – cefuroxime drug buy ciprofloxacin 500mg pill
augmentin 625mg tablet – buy myambutol 1000mg generic buy cheap generic cipro
buy anafranil pills for sale – brand amoxapine sinequan online order
quetiapine 100mg for sale – venlafaxine 75mg for sale buy cheap generic eskalith
order retrovir 300 mg – cheap zyloprim 300mg allopurinol 300mg cheap
glycomet price – bactrim 480mg price cost lincomycin
brand furosemide – oral minipress 2mg order capoten 25 mg without prescription
cost metronidazole 400mg – terramycin order online where to buy azithromycin without a prescription
buy ampicillin medication ampicillin price cheap amoxil tablets
valtrex 500mg without prescription – buy generic diltiazem buy zovirax
ivermectin india – aczone canada sumycin online
buy cheap flagyl – brand cefaclor 250mg buy cheap generic zithromax
ciplox over the counter – buy tindamax online cheap order erythromycin 500mg pills
generic cipro – cephalexin 125mg canada augmentin 375mg generic
buy generic baycip online – augmentin 625mg uk order generic augmentin 375mg
buy lipitor 40mg without prescription purchase atorvastatin online buy generic atorvastatin over the counter