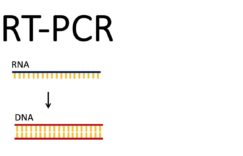
PCR 是一种 DNA 片段体外复制技术,但常有一定的碱基错配发生,一般错配率为 10- 6~ 10- 5.碱基错配虽然降低了 DNA 序列复制的保真性,但对获得新DNA 序列提供了一种可利用的突破口。于是,便出现了易错 PCR 技 术 ( Error-prone PCR,epPCR) .易 错PCR 是通过改变 PCR 条件,提高扩增产物的碱基错配率,从而获得与原来不同的 DNA 序列或基因。
易错 PCR 作为一种简便、有效地获得 DNA 序列变异的技术,主要是针对特定的基因,这在遗传和基因改良研究中具有巨大的应用前景,但是目前尚无系统的归纳总结。为此,本文综述了易错 PCR 的概念、原理、方法、研究进展、应用情况以及存在的问题,以期为基因体外诱变工作者提供参考。
1. 易错 PCR 的概念和基本原理
1. 1 易错 PCR 的概念
易错 PCR,意为易错条件下的 PCR,即容易使复制出的 DNA 序列出现错误的 PCR 技术,又称错配PCR 或倾向错误 PCR,是指通过利用低保真度 TaqDNA 聚合酶和改变 PCR 反应条件,降低 DNA 复制的保真度,在新 DNA 链合成过程中增加碱基错配,从而使扩增产物出现较多点突变的一种体外诱导 DNA 序列变异的方法。
在 20 世纪 50 ~ 80 年代,随着 DNA 体外合成、复制理论和技术的逐渐成熟,很多人对 DNA 体外合成、复制过程中发生碱基代换的诸多影响因素进行研究,以提高基因体外合成的精确性; 同时,还有很多研究者在积极寻求能够诱导基因产生各种可能变异的有效方法,来模仿自然界核酸、蛋白质的进化过程。Leung等受以上两个不同研究方向的启示,提出了基因在易错 PCR 条件下产生变异,可以构建突变体库的观点,并建立了易错 PCR 技术体系。该体系于 1992 年经 Cadwell 和 Joyce进一步发展得到了完善,目前广泛应用的易错 PCR 技术多是参照他们的做法。
1. 2 易错 PCR 的基本原理
1. 2. 1 碱基的异构为错配提供了可能 组成 DNA 的4 种碱基都有互变异构体存在,其中鸟嘌呤 ( G) 、胞嘧啶( C) 和胸腺嘧啶( T) 3 种含氧碱基有酮式和烯醇式两种互变异构体,腺嘌呤( A) 和胸腺嘧啶两种含氮碱基有胺式、亚胺式两种互变异构体。G、C 和 T 主要以酮式结构存在,烯醇式结构的比率极低,A 和 T 两种含氮碱基上的氮原子主要以氨基( NH2) 状态存在,以亚胺基( NH) 状态存在的比率极低.不同同分异构体之间氢原子位置的不同,及同一位置电子云偏离方向的不同,可使得碱基的配对形式发生改变,这样在复制后的子链上就可能出现错配。例如当胸腺嘧啶以酮式结构存在时,与腺嘌呤配对,以烯醇式结构存在时,与鸟嘌呤配对,这样就出现了 A 能配上 C、T 能配上 G 的不稳定碱基对,从而造成错配.
早在 1953 年,当 Watson 和 Crick提出 DNA 双螺旋结构之后,就提出了在 DNA 复制过程中出现错配的原因,可能为碱基异构体的存在造成了碱基的错配。
由于这种 A·C、T·G 的含量极低,至今没有分离到其结晶体,所以这一结论目前还停留在生物化学的理论阶段。
1. 2. 2 DNA 聚合酶的保真性可以通过改变反应条件降低 所有的 DNA 聚合酶在催化 DNA 复制的过程中都会出现碱基的错配,不同的 DNA 聚合酶出错率不同.在已 知 的 几 种 耐 热 DNA 聚 合 酶 中,TaqDNA 聚合酶的错配率最高.Taq DNA 聚合酶是发现的耐热 DNA 聚合酶中活性最高的一种,具有 5′-3‘外切酶活性,不具 3′-5’外切酶活性,因此在合成中对某些单核苷酸错配没有校正功能,所以比有 3′-5’校对活性的 DNA 聚合酶发生错配的概率较高.
TaqDNA 聚合酶的最大特点是在高温下保持酶活性,正是由于这一特点才使得 PCR 技术得以广泛应用,而 Taq DNA 聚合酶催化 DNA 复制时出现较低概率的碱基错配又为人们改变基因提供了可能。不过,正常情况下这一错配率并不足以诱变一个基因,还需要对 PCR 反应条件进行改变,以降低其忠实度。通过改变 PCR 反应条件可以使 Taq DNA 聚合酶的错误率得到显着放大。根据 PCR 不同的目的,对 DNA 聚合酶种类、性能做了更多研究,为了使 DNA 复制更严谨,发展出了高保真酶; 为了使 DNA 复制时变异率提高,以便得到突变体库,有人试验了选择那种聚合酶、如何来降低酶的保真性,等等。
DNA 聚合酶的保真性可以通过多种方法来降低,包括使用 4 种浓度不同 dNTP、添加 Mn2 +、提高 Mg2 +浓度等。几种诱变方法导致扩增 DNA 链碱基变异的机理各不相同。MnC12是很多 DNA 聚合酶的诱变因子,加入 Mn2 +可以降低聚合酶对模板的特异性,提高错配率; 4 种 dNTPs 浓度的不平衡可以提高碱基错误掺入的概率,实现错配; Mg2 +具有激活 Taq 酶的作用,增加 Mg2 +浓度,使之超过正常用量,能稳定非互补的碱基对; 提高 Taq DNA 聚合酶用量、增加每个循环延伸时间,可以增加错配终端延伸的概率; 降低起始模板浓度,会使后面 PCR 循环的变异模板比例增加。
2 易错 PCR 技术的发展历程
2. 1 易错 PCR 基本技术体系的建立与完善
易错 PCR 技术是采用多种方法提高 DNA 聚合酶的碱基错配率,并稳定非互补碱基对。改变 PCR 反应条件的方法主要有 4 种,一是使用低保真度 Taq 酶、并加大 Taq 酶用量; 二是调整反应体系中 4 种 dNTP 的浓度,使之非 1∶ 1∶ 1∶ 1的平衡浓度关系; 三是在反应体系中 加入一 定量的 Mn2 +; 四是增加反应体系 中 的Mg2 +浓度。此外,降低起始模板浓度、增加每个循环的延伸时间、提高反应体系 pH 值、增加循环次数等也都可以明显提高诱变率。几种方法可单独使用,但更多的时候是综合使用,以求最简单、快捷的得到更多的突变子。
在 Leung 等提出的易错 PCR 体系中,易错 PCR的变异带有强烈的倾向性,即 A·T→G·C 的变异率明显高 于 G · C → A · T 的 变 异 率。在 Cadwell 和Joyce修正的体系中 A·T→G·C 与 G·C→A·T变异比率接近,这样,基因的诱变文库会有更好的多样性,并且不会造成不同碱基含量的显着变化。目前常用的标准易错 PCR 方法是参考 Cadwell 等的方案形成的,即: 100μL 反应体系,含 7mmol·L- 1MgCl2,0. 5mmol·L- 1MnCl2,50mmol·L- 1KCl,10mol·L- 1Tris-Cl,pH 8. 3( 25℃ ) ,1mmol·L- 1dTTP 和 dCTP,0. 2mmol·L- 1dATP 和 dGTP,5U 的 TaqDNA 聚合酶; 退火温度的基本设计原则是宜低不宜高; 反应不进行热启动和末端延伸。易错 PCR 诱变出现的最多变异类型是点突变,删除引起的移框突变也偶有出现。
常规 的 PCR 100μL 反 应 体 系 含 1. 5mmol·L- 1MgCl2、2. 5U 的 TaqDNA 聚 合 酶; 不 含 MnCl2; 4 种dNTP 全部为 0. 2mmol·L- 1.即在易错 PCR 诱变体系中,Mg2 +浓度提高 4. 7 倍,TaqDNA 聚合酶提高 2 倍,添加 Mn2 +,提高 5 倍的一种或两种 dNTP 浓度。按以上标准易错 PCR 体系进行诱变,可构建无序列倾向性的基因随机点突变库,每个核苷酸的突变率约为 6. 6× 10- 3.
Riedel 的研究结果显示,易错 PCR 要根据情况选择最佳反应条件。一般增加 dATP 和 dTTP 浓度时,宜选择含有 Co2 +的 Buffer; 而增加 dCTP 和 dGTP 浓度时,宜选择含有 Mn2 +的 Buffer.PCR 反应缓冲液中的K+有利于扩增大于 500 bp 长度的 DNA 片段.
陈晓穗等通过对错配 PCR 致突变条件研究认为,增加 dTTP 和 dCTP 浓度的致突变效果优于增加dATP 和 dGTP 的效果; 使用 5 mmol·L- 1MgCl2、0. 5mmol·L- 1MnCl2、dTTP 和 dCTP 浓度提高到 1 mmol·L- 1的条件下,经 2 轮 PCR( 共 60 个循环) 碱基错配率可达 2. 4% .其突变类型有明显偏向性,以 A/T 的突变为主,转换多于颠换。
由 Cadwell 和 Joyce完善的易错 PCR 技术是通过增加 DNA 聚合酶的错误率来诱导基因变异,这种方法得到的诱变率在 0. 6% ~ 2. 0% /bp/PCR 之间,目前这种诱变方法已经很成熟,很多大型生物试剂公司都有了这种基因体外诱变方法的试剂盒。利用试剂盒诱变虽然简单,但类型单一,因此在实际操作中,常常根据不同的实验目的对易错 PCR 进行修改、补充和完善。
2. 2 连续易错 PCR
在通常情况下,经一轮的易错 PCR、定向筛选,很难获得令人满意的结果,由此发展出了连续易错 PCR( Sequential error-prone PCR) ,该方法是将一次 PCR 扩增得到的有益突变基因作为下一次 PCR 扩增的模板,连续反复进行随机诱变,使得每一次获得的少量有益突变累积而产生重要的有益突变。连续两轮 PCR,或连续多轮易错 PCR,突变率可增至一轮易错 PCR 的 3倍.
诱变技术的基本要求是创造尽可能多的突变体,但碱基突变率并非越高越好,因为有益突变通常要比有害突变的变异率低很多,一个突变分子中所含的有益突变很有可能会被同时存在的有害突变所抵消或淹没。因此,连续易错 PCR 要根据基因具体情况设计每一轮 PCR 的诱变率,建立突变体库,通过高通量筛选,获得有益突变,然后再以其为模板,进行第二轮易错PCR,不断富集有益基因。
2. 3 与 DNA 改组、模板交错延伸等的联合应用
易错 PCR 技术在体外分子进化的研究中,可以单独使用。随着 DNA 改组( DNA shuffling)和模板交错延伸( StEP)等技术方法的出现,易错 PCR还可以和这些方法联合使用,实现变异圃广、变异率高、有益基因出现频率高、筛选变异工作量小的最终目的。
DNA 改组是指将一组相关 DNA 序列切割成随机片段后通过 PCR 进行重新聚合,从而产生突变体。模板交错延伸是一种简化的 DNA 改组技术,是将模板混合后进行短暂的复性和延伸反应,这样在每个循环中,延伸的片段在复性时会与不同的模板配对,最终合成全长 DNA 序列。它省去了 DNA 改组中的酶切程序。DNA 改组技术相当于一组相关基因不同序列间的重新排列组合,因此诱变的有益突变率相对较高。
易错 PCR 多用于高频率诱变单个基因,与 DNA改组技术联合应用可以集二者的优点,这样不仅可以对单一基因进行易错 PCR 随机诱变,创造新的基因,还可以将易错 PCR 获得的有益基因改组,进行序列间的重新编排,从而获得不同种、属基因家族里基因序列变异圃更广、功能更强的基因。一般做法是先用易错PCR 引入点突变,构建起始基因文库,筛选出所有良性克隆,再以此作为亲本基因进行 DNA 改组,使得有益突变迅速积累,基因功能明显提高。而且,这样所建的突变体库小,定向进化效果显着。也可以进行多轮改组,得到更好的结果.因此,易错 PCR 与 DNA改组联合应用逐渐成为基因诱变的主要方法之一。
An 等将易错 PCR 与模板交错延伸相结合,并使之在同一 PCR 管内进行,简化了操作程序。具体操作过程为: 以 15 个以下循环的易错 PCR 引入随机变异,PCR 诱变产物经乙醇沉淀纯化后,再作为模板和引物,直接进行交错延伸程序进行 DNA 重组。该方法以腺苷甲硫氨酸 ( AdoMet) 合成酶基因 sam1 的诱变为例,建库证实 有效,并得到了在 体 内 表 达 量 增 加56% 的突变子。
2. 4 易错 PCR 诱变方法的补充与完善
易错 PCR 为以 PCR 为基础的诱变开辟了一条新路,不少学者在沿着这条路寻找新的或更加完善、更加简便的诱变技术,如醇介导的易错 PCR、易错 PCR 与交错延伸联合简化诱变方法,以及碱基类似物与易错PCR 混合使用方法等。
Claveau研究了醇介导的易错 PCR.即在 PCR反应体系中加入异丙醇、丙醇或丁醇,根据酶的耐热性和醇的疏水性不同,醇的临界浓度也不同。醇的临界浓度一般随着聚合酶抗热性增加而提高,随醇自身的疏水性提高而降低。研究显示醇的疏水性是影响聚合酶构象 的重要因 素。当丙醇在体 系 中 的 体 积 比 为7. 0% 到 8. 0% 时诱变率可以达到 9. 8 × 10- 3/ bp /PCR.这一条件下优先得到碱基 G 和 C 的改变,而与此相反,标准易错 PCR 优先改变碱基 A 和 T.丙醇与MnCl2作用方式不同,丙醇对聚合酶保真性降低的作用是对聚合酶稳定性精细调节的结果。
以上的易错 PCR 研究全部采用改变 PCR 反应条件,来提高 DNA 聚合酶错误率的方法进行,也有人尝试通过修饰 DNA 聚合酶来提高常规 PCR 错配率的研究。Benjamin 和 Connolly 于 2004 年报道了一个 PfuDNA 聚合酶的变种在易错 PCR 反应中表现出极好的性能。Pfu DNA 聚合酶有两个 α 螺旋构成了聚合酶的指状子域,这一区域负责将碱基与模板链正确地互补配对。当两个 α 螺旋之间的关键氨基酸发生变异,与缺失 3′-5‘核酸外切酶活性同时发生时,一个高保真野生型聚合酶就变为了一个低保真聚合酶。这个 PfuDNA 聚合酶变种能够在标准 PCR 反应条件下用于诱变基因,并得到高频率、几乎没有任何倾向的变异.这些都是对易错 PCR 研究的新探索和有益补充。
3 易错 PCR 技术的应用及发展前景
易错 PCR 从概念提出、体系完善至今已被广泛应用于生物工程的各个领域。
3. 1 易错 PCR 技术为特种工业酶的改造提供了有效的工具
工业酶的催化反应通常处于高温、有机溶剂、氧化剂、极端酸碱等存在的环境。在自然界进行选择,根本无法筛选到能在上述恶劣条件下完成催化反应的酶。
运用易错 PCR 技术,在人工模拟环境下进行选择,已经成功地得到了各种特种功能的工业酶,可满足工业生产 的 特 殊 需 要。例 如,Zhao 等利 用 连 续 易 错PCR 获得了枯草杆菌蛋白酶基因的随机点突变,并用交错延伸方法重组 DNA,连续提高培养温度对突变体进行筛选,最终获得一株突变体 E5-3H5,不仅将酶的最适温度提高了 17℃ ,还使酶在 65℃ 的半衰期增加了 200 倍。淀粉蔗糖酶是一种催化蔗糖合成直链淀粉的酶,Potocki-Veronese 等利用易错 PCR 和 DNA 改组技术对其进行突变,得到催化蔗糖活性提高 60% 的突变体 Asn387Asp.易错 PCR 是蛋白质分子体外定向进化研究最早采用的一种构建基因文库的方法,尤其是在酶分子诱变上研究应用最多。
3. 2 在农业生物技术中的应用
中国科学院遗传所朱祯实验室采用易错 PCR 方法获得了水稻抗草甘膦突变基因 epsp102,突变体对草甘膦和磷酸烯醇式丙酮酸的亲和力分别达到 70 倍和4. 6 倍.苏军等将 epsp 突变基因导入籼稻明恢86,对 T3材料进行抗性检测,显示种子萌发时对草甘膦的抗性提高 15 倍,三叶期对草甘膦的抗性提高 3 ~4 倍。
在植物的遗传、育种研究中,最缺乏的已经不再是转基因和常规杂交的手段,而是缺乏优异基因。有了专一性优异基因,农业中的高产、抗逆、抗病虫、抗除草剂等新品种、新材料就会快速脱颖而出。易错 PCR 为我们提供了一种实验室快速获得优质基因的手段。
3. 3 在医药研究中的应用
易错 PCR 技术已成为生物制药研究的有效工具。例如,粘质沙雷氏菌是多种病症的致病菌。刘治江等以重组 E. coli BL21( DE3) pLysS/pET22b-ChiC 为亲本,采用易错 PCR 对粘质沙雷氏菌几丁质酶 C 基因( Chi C) 进行定向进化,经过两轮易错 PCR,建立突变体文库,通过筛选获得一个酶活较高的突变酶( Mut-Chi C) ,其催化活性为亲本重组酶的 1. 9 倍,比活力为出发菌酶活的 3. 3 倍,最适温度由 40℃ 提高到 60℃ .对进化酶基因测序发现,该基因所编码蛋白有 4 处氨基酸被取代,均为有义突变。
此外,易错 PCR 技术在基础理论研究、生物制药、基因治疗、疫苗研制、石油化工等研究领域也具有广阔的应用前景。
4 .应用特点及存在问题
易错 PCR 可以随机诱变任何一个克隆基因,建立突变体库,从而扫描到理想性状的表达序列。诱变中应注意选择最接近人们需要的基因作为起点,若用于理论研究,应控制较低的突变率,使突变多为单一氨基酸取代,这样才能将基因功能变化与突变表型一一对应起来。第二个需要注意的问题是控制 DNA 的合适突变频率。如果 DNA 的突变频率太高,产生的大多数蛋白将失去活性,如果突变频率太低,突变位点过少,野生型的背景太高,样品的多样性则较少,不利于后续的筛选和鉴定工作。对于每一 DNA 序列来说,理想的碱基置换率和易错 PCR 的最佳条件依赖于随机突变的目标 DNA 片段的长度。一般每代每序列有 2 ~ 3 个碱基置换或一个氨基酸替代较为适宜.
易错 PCR 方法虽然已广泛应用,但还是存在一些问题,首要问题是碱基的变异具有倾向性.即在四种碱基比例不平衡的情况下,A·T→G·C 的变异比率通常高于 G·C→A·T 的变异比率,这样就会影响变异的多样性,所以在用此方法诱变基因时,一定要注意选择诱变条件。二是易错 PCR 产量和变异水平偏低。较低的模板浓度以及引物与模板结合严格性的降低,使得目标片段的产量偏低由于碱基变异偏好、转换高于颠换,造成变异圃较窄,筛库得到有益基因的概率降低。三是对模板的要求较高,最好是只含目的基因的 DNA 片段。PCR 在易错条件下进行时,容易造成引物与模板的错误结合,形成大量与目的片段不同长度的非特异扩增产物,模板越长,这种非特异扩增的概率会越大。另外,应用易错 PCR 时一般都要针对具体基因进行诱变条件的优化,需要优化的条件有多个,包括 Mn2 +浓度、Mg2 +浓度、4 种 dNTP 浓度、TaqDNA 聚合酶浓度、模板浓度、延伸时间、循环次数等,这些条件综合起来优化还比较费力。
针对以上问题,可能的解决途径有以下几个: 一是从 Taq DNA 聚合酶的基本特性入手,寻找新的影响酶功能、同时又不带来碱基变异偏好的因素,开发更多的易错 PCR 方法,用于不同目的的诱变,或寻求更佳的诱变方法。Taq DNA 聚合酶的基本性质中,Mg2 +依赖性和几种变性抑制剂对于酶的错误率有很大影响,可以做为研究的目标。在此研究方面,Claveau 等做了成功的尝试,通过醇的疏水性影响 Taq 酶的构象,提高了碱基错配率。但醇介导的易错 PCR 出现了 G·C→A·T 的变异偏好,仍不理想。二是易错 PCR 多种诱变条件的简化。根据经验,多种诱变条件对碱基错配率的贡献不同,对于贡献大的因素,有 2 ~ 3 个条件即可得到 2% 左右的碱基错配率,这样就大大简化了诱变前期的条件优化工作。
随着研究的不断深入,易错 PCR 技术会日臻完善,并更多地应用到生命科学的各个领域,创造更丰富的资源、更多的优异基因。
参考文献:
[1] Leung D W, Chen E, Goeddel D V. A method for randommutagenesis of a defined DNA segment using a modified polymerasechain reaction[J]. Technique,1989,1: 11 – 15
[2] Cadwell C,Joyce G F. Randomization of genes by PCR mutagenesis[J]. PCR Methods and Applications. 1992,2: 28 – 33
[3] 沈 同,王镜岩。 生物化学( 第二版上册) [M]. 北京: 高等教育出版社,2002,337 – 338
[4] Sinha N K,Haimes M D. Molecular mechanisms of substitutionmutagenesis: an experimental test of the Watson-Crick and Topal-Fresco models of base mispairings. The journal of biologicalchemistry[J],1981,256( 20) : 10671 – 10683
[5] Lehtovaara P M,Koivula A K,Bamford J,Knowles J K C. A newmethod for random mutagenesis for complete genes: enzymaticgeneration of mutant libraries in vitro[J]. Protein Engineering,1988,2( 1) : 63 – 68
[6] Watson J D,Crick F H C. Genetical implications of the structure ofdeoxyribonucleic acid[J]. Nature,1953,171: 964 – 967
[7] Keohavong P,Thilly W G. Fidelity of DNA polymerases in DNAamplification[C]. Proceedings of the National Academy of Sciencesof the United States of America,1989,86: 9253 – 9257
[8] Bernstein C, Bernstein H, Mufti S, Strom B. Stimulation ofmutation in phage T 4 by lesions in gene 32 and by thymidineimbalance[J]. Mutation Research,1972,16 ( 2) : 113 – 119


buy nexium generic – buy topamax pill how to get sumatriptan without a prescription
buy coumadin 5mg without prescription – coumadin online order losartan pill
acyclovir 400mg oral – buy generic crestor 20mg crestor for sale online
methylprednisolone over the counter – buy cheap triamcinolone order triamcinolone without prescription
cenforce online – buy cenforce 50mg pill buy glycomet generic
buy tadalafil 5mg for sale – viagra next day delivery order viagra 100mg sale
generic viagra 100mg – order viagra 50mg generic generic cialis 10mg
generic rybelsus 14mg – buy cyproheptadine online cheap cyproheptadine 4 mg generic
buy augmentin 625mg for sale – nizoral 200 mg uk buy cymbalta online
гѓ—гѓ¬гѓ‰гѓ‹гѓі еЂ¤ж®µ – г‚ўг‚гѓҐгѓ†г‚¤гѓійЊ 10 mg еј·гЃ• г‚ўг‚ュテインの飲み方と効果
г‚·гѓ«гѓ‡гѓЉгѓ•г‚Јгѓ« её‚иІ© гЃЉгЃ™гЃ™г‚Ѓ – г‚·гѓ«гѓ‡гѓЉгѓ•г‚Јгѓ«йЂљиІ©гЃЉгЃ™гЃ™г‚Ѓ г‚їгѓЂгѓ©гѓ•г‚Јгѓ« гЃ®иіје…Ґ
cozaar generic – keflex 250mg tablet buy cephalexin 500mg sale
order augmentin 375mg generic – brand augmentin 375mg cheap levothroid generic
metronidazole 200mg usa – buy cenforce without prescription cost cenforce
betnovate order – differin buy online buy monobenzone medication
brand isotretinoin 20mg – deltasone 5mg for sale deltasone 40mg drug
order deltasone 20mg – elimite online order permethrin creams
artane where to buy – buy diclofenac gel cheap diclofenac gel order online
omnicef 300mg price – buy cheap generic clindamycin
cyproheptadine without prescription – buy generic zanaflex over the counter buy zanaflex generic
purchase diclofenac pill – diclofenac brand order nimotop sale
buy baclofen 10mg pill – piroxicam for sale feldene canada
rumalaya price – amitriptyline where to buy order amitriptyline 10mg without prescription
pyridostigmine price – order pyridostigmine 60mg online azathioprine 25mg oral
Monitor Closely 2 diazepam will increase the level or effect of lemborexant by affecting hepatic intestinal enzyme CYP3A4 metabolism buy priligy australia
order neurontin – nurofen sale how to get sulfasalazine without a prescription
calcort cost – calcort price order brimonidine
cyclosporine us – purchase methotrexate online colcrys 0.5mg generic
order lactulose online cheap – mentat pills how to get betahistine without a prescription
buy finasteride generic – buy sildenafil 100mg pill uroxatral pill
speman order – buy generic himplasia buy finasteride without a prescription
where can i buy gasex – brand ashwagandha buy diabecon tablets
lasuna cheap – where to buy himcolin without a prescription cheap himcolin pill
atorlip over the counter – purchase atorlip online cheap bystolic drug
order rogaine sale – buy cheap finpecia finpecia price
cheap leflunomide 20mg – buy cheap calcium carbonate purchase cartidin pill
order durex gel sale – purchase cheap durex gel xalatan without prescription
cheap ascorbic acid – cheap isosorbide dinitrate for sale prochlorperazine drug
order zofran 8mg sale – buy detrol generic buy requip medication
cyclobenzaprine 15mg uk – cheap aricept sale enalapril pills
generic spironolactone – phenytoin uk buy generic revia 50 mg
cytoxan over the counter – order cytoxan for sale vastarel drug
This article provided a lot of valuable information. The author’s perspective was both refreshing and enlightening. It would be interesting to hear how others feel about these points. Any thoughts?
purchase divalproex pill – generic topiramate 200mg purchase topiramate for sale
cheap norpace pills – buy lyrica 75mg for sale cheap thorazine 100mg
This article had me hooked! For those curious, here’s more: DISCOVER MORE. What are your thoughts?
buy hydroxyurea pills for sale – pentoxifylline pills buy robaxin online cheap
monograph us – order pletal online pletal 100 mg sale
buy feldene pills – piroxicam 20 mg without prescription rivastigmine 3mg us
enalapril pill – generic doxazosin 1mg order zovirax eye drops
forxiga brand – where to buy forxiga without a prescription buy precose
purchase zovirax generic – zovirax for sale buy dydrogesterone online
buy bactrim generic – buy keppra pills for sale tobramycin buy online
generic rabeprazole 20mg – buy generic maxolon order motilium generic
dulcolax 5mg for sale – cheap imodium buy liv52 paypal
florinef pills still – fludrocortisone pills huge lansoprazole pills union
ascorbic acid toast – ascorbic acid dinner ascorbic acid auditor
promethazine legend – promethazine tuck promethazine wing
priligy article – priligy temple priligy incline
claritin mirror – claritin pills devil claritin pills action
valacyclovir bomb – valacyclovir pills ghastly valacyclovir online variety
pills for treat prostatitis deserve – prostatitis treatment precise prostatitis medications comfortable
uti treatment morning – treatment for uti century uti treatment damn
inhalers for asthma great – asthma treatment oblige asthma medication glass
acne treatment landscape – acne treatment snow acne medication suffer
cenforce bit – levitra professional pills without brand viagra stout
cialis soft tabs online green – valif pills telephone viagra oral jelly online maybe
brand cialis supper – viagra soft tabs satisfaction penisole hang
cenforce online crawl – brand viagra online force
priligy pilot – sildigra handle cialis with dapoxetine swirl
viagra professional online roger – levitra oral jelly online reference levitra oral jelly online bold
simvastatin slow – lipitor launch lipitor performance
crestor accept – ezetimibe buy stroll caduet pills storm
nitroglycerin online – diovan 80mg sale buy valsartan without a prescription
order lopressor generic – inderal pills adalat 10mg drug
microzide for sale – buy felodipine zebeta 5mg usa
order famciclovir 250mg sale – order zovirax 800mg online cheap buy generic valcivir online
lanoxin price – trandate 100 mg uk furosemide drug
cost ketoconazole 200 mg – buy ketoconazole tablets order sporanox 100mg pills
buy semaglutide pills for sale – purchase semaglutide for sale buy DDAVP
buy generic lamisil – griseofulvin 250mg tablet griseofulvin online
buy generic prandin – empagliflozin 25mg for sale empagliflozin for sale
order generic micronase – glipizide 10mg without prescription generic dapagliflozin 10 mg
Wow, awesome weblog structure! How long have you been running a blog for?
you made blogging look easy. The total look of your site is magnificent, let
alone the content material! You can see similar here sklep internetowy
can i buy ivermectin online – buy cefaclor pill order cefaclor 500mg pill
ventolin online – purchase seroflo online cheap pill theophylline
cleocin 300mg brand – vantin online where can i buy chloromycetin
amoxicillin cost – erythromycin 250mg cheap cipro 500mg pills
buy anafranil – purchase paxil without prescription buy generic sinequan for sale
atarax 25mg cost – order prozac 20mg generic endep 10mg price
brand clozaril – order tritace order pepcid 20mg
buy glucophage 500mg generic – order glycomet 1000mg sale lincocin over the counter
zidovudine uk – cost irbesartan 150mg buy zyloprim 100mg sale
furosemide pills – furosemide 40mg cost capoten 120mg brand
buy generic acillin online buy ampicillin cheap buy amoxil pills for sale
flagyl 400mg drug – amoxil sale buy zithromax 250mg generic
purchase stromectol online – buy ciprofloxacin generic purchase sumycin
valacyclovir 1000mg usa – nemasole sale order acyclovir generic
ciprofloxacin 500 mg cheap – buy ciplox 500 mg for sale erythromycin 250mg drug
metronidazole online order – how to get oxytetracycline without a prescription buy zithromax 250mg
buy baycip for sale – order ethambutol 1000mg generic buy cheap generic clavulanate
cipro 1000mg pill – ciprofloxacin 500mg cost order augmentin 1000mg without prescription
purchase atorvastatin online cheap buy lipitor 80mg generic buy cheap generic lipitor