PCR,相信我们都不陌生,是一种选择性体外扩增DNA或RNA片段的方法,即通过试管中进行的DNA复制反应,使极少量的基因组DNA或RNA样品中特定基因片段在短短几小时内扩增上百万倍。
简单说,就是我们在体外模仿体内DNA复制的过程,混合DNA模板、合适的引物、足够的4种dNTP、耐热DNA聚合酶以及适宜的体系后,在一定温度和时间条件下,进行的DNA扩增。
PCR有很多种,其中RT-PCR,全称反转录聚合酶链反应(reverse transcription-polymerase chain reaction, RT-PCR)是将RNA的反转录和cDNA的聚合酶链式扩增(PCR)相结合的技术。
实验原理是在试管内,以变性-退火-延伸三个基本反应为一个循环,反复重复这种循环,使DNA得以扩增。其目的是检测核酸的表达,主要是RNA的表达,DNA不需要反转录的过程,直接PCR检测即可。
一、RT-PCR操作步骤
1. RNA抽提
方法:TRlzol法。
主要成分:苯酚(具有裂解细胞和变性蛋白的作用)。
RNA抽提前准备:试剂耗材准备(注意RNase free)、样品的准备。
步骤:样品+1ml TRlzol室温裂解10min+200μL氯仿振荡30s后室温静置2-3 min;4℃低温离心(12 000g×20min);400μL上层水相+600μL异丙醇室温沉淀10min;4℃低温离心(12 000g×20min);75%乙醇冲洗;晾干沉淀,DEPC-treated水溶解。
2. RNA 质量检测
方法:分光光度计测定、凝胶电泳法
3. 反转录
是以RNA为模板合成cDNA的过程。在这个过程中遗传信息的流动方向和转录时相反,所以称为“反转录”。在这个过程中需要一个反转录酶,合成的DNA称为与RNA互补的DNA(complementary DNA, cDNA)。这个过程中根据不同的情况选择不同的反转录引物,如果要检测的RNA有poly A尾巴,可以用Oligo(dT)做反转录引物;如果要检测的RNA没有poly A尾巴,就用随机引物(random primer)来反转;如果要检测的RNA序列已知,可用特定引物(specific primer)来反转。
4. PCR程序
三步法:
第一步:94℃变性30s;
第二步(循环30-35次):94℃变性30s,55-60℃退火30s,72℃延伸24s(时间按照合成速度和长度来计算);
第三步:72℃终止延伸5-10min。
5. 引物
引物设计步骤:
找到目的基因的CDs序列(存在多个转录本时可对各个转录本的保守区域进行引物设计);用软件设计和评价引物(Oligo软件);blast引物特异性。
要求:
引物长度为15-30bp,最常用的为23bp;引物Tm值介于62-73℃之间,上下游引物Tm最好相近以保证其能高效工作;3’-端的Tm值要低于5’端;引物GC含量约为40-60%;避免扩增模板的二级结构域;避免引物的二级结构;避免与靶DNA错配。
6. PCR酶:可按需要选择普通的Taq酶、可扩增长片段的Taq酶或高保真酶。
7. PCR体系:按所选的PCR酶说明书来配制。
8. PCR产物检测:一般采用琼脂糖凝胶电泳,必要时也可使用聚丙烯酰胺凝胶电泳。
反转录引物选择:
1. Oligo dT:适用于具有PolyA尾巴的RNA。(原核生物的RNA、真核生物的rRNA和tRNA不具有PolyA尾巴。)由于Oligo dT要结合到PolyA尾巴上,所以对RNA样品的质量要求较高,即使有少量降解也会使全长cDNA合成量大大减少。
2. 随机引物(random primer): 适用于长的或具有发卡结构的RNA。适用于rRNA、mRNA、tRNA 等所有RNA的反转录反应。
3. 特异性引物(Specific primer):与模板序列互补的引物,适用于目的序列已知的情况。
二、RT-PCR需注意的细节
1. 实验过程中必须戴口罩、手套,尽量避免其他人员干扰。
2. RT试剂盒从冰箱拿出,应该充分解冻,瞬离后,混匀,放于冰上,可在冰上加样。试剂可以用数字贴纸编号,避免漏加、错加试剂。
3. 取用不同的试剂时,必须更换枪头,样本之间要避免交叉污染。
4. 逆转录时,PCR仪可以先预热。
5. 反应体系配好之后,应充分混匀,瞬离,并且弹去管内的气泡。
6. 实验过程中应预防RNA酶污染,使用无酶枪头以及EP管,同时台面可以使用固相RNA酶去除剂清洁。
7. PCR时,管壁尽量避免写字。
8. 管盖一定要盖严。
9. 10μL逆转录反应体系建议加入不超过500ng的RNA。
10. 如果目的基因表达量非常低,最多加入1μg Total RNA,否则加入RNA量过高,可能会超出后续PCR的线性范围。
11. 反应体积可根据需要等比例放大。
12. 试剂尽量不要反复冻融。
13. 每个样品至少3个平行孔,所有成分加完后,离心去除气泡。
14. 可将所有共同物质加入同一管中,混匀后分散入其他管中。
三、RT-PCR写作例句
第一句
Total RNA was extracted with/ isolated from cultured cells/ samples/ tumor tissues using/ by Trizol reagent (Invitrogen, Thermo Fisher Scientific, Braunschweig, Germany)/ RNeasy kit (Qiagen)/ RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol /following the manufacturer’s (recommended) instructions as described previously.
TRIzol reagent (Thermo Fisher Scientific) was used to extract RNA from samples.
第二句(可与第一句合并)
RNA (2 μg) was converted into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo).
Total RNA was reverse-transcribed into cDNA using the aScript cDNA SuperMix (Quanta Biosciences).
2 μg RNA was used to synthesize cDNA using the PrimeScript RTreagent kit (Takara, DRR037A).
第三句
Gene primers (…) were designed using Primer Express v3.0 Software.
The primer sequences used are listed/shown in Supplemental Experimental Procedures/ Table X. 也可写与句末。
The following primers were used:/ Gene-specific primers as follows: Gene名称forward 5′-XXX-3′ and reverse 5′- XXX-3′;
第四句
RT-PCR was performed with/ accomplished using/ carried out using …according to the manufacturer’s instructions.
第五句
GADPH /U6 was used as endogenous controls/ an internal reference. Relative expression level was computed using 2-ΔΔCt method.
For the data of each sample, the Ct value of the target gene was normalized to that of U6 (…).
The expression level of each gene was performed and the data were normalized to control unwanted sources of variation.
The relative expression level of indicated genes was compared with that of… and expression fold changes were calculated using 2-△△Ct methods.
第六句(可写可不写)
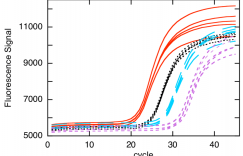
Each qRT-PCR reaction was performed in triplicate as follows: step 1: denaturation at 95°C for 10 min; step 2: 40 cycles of 95°C for 15 s and 60°C for 1 min.
PCRs were a relative estimation in triplicate as per the following temperature profile: denaturation 95 °C for 10 min followed by amplification by 40 cycles of 95 °C for 10 s and 60 °C for 1 min.
第七句(可写可不写)The cDNA was then diluted to 70 μL in water. qPCR was performed with 2×SensiFAST SYBR No-ROX kit (Bioline), gene-specific primers (250 nM final concentration of forward and reverse), and 2 μL cDNA.
举例
1. Total RNA was extracted using a Trizol reagent (Invitrogen). RNA (2 μg) was converted into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo). QRT-PCR was accomplished using the FastStart Universal SYBR Green Master (Rox) (Roche) in the ABI PRISM® 7300 real-time PCR system (Applied Biosystems, Foster City, CA, USA). GADPH and U6 were used as endogenous controls. We used melting curves to monitor non-specific amplifications. Relative expression level was computed using 2-ΔΔCt method. The primer sequences used are listed in Supplemental Experimental Procedures.
2. Total RNA was extracted by TRIzol reagent as described previously xx. The expression levels of 7 randomly selected differentially expressing circRNAs (Fold changes ≥ 2, p < 0.05) were measured by qRT-PCR; among them, 2 were upregulated and 5 were downregulated in the GC tissues: (upregulated: hsa_circ_0081146, hsa_circ_0084720), (downregulated: hsa_circ_0060108, hsa_circ_0057104, hsa_circ_0054971, hsa_circ_0063561, and hsa_circ_0058766). GAPDH expression was used as an internal reference. The primers used for these amplifications are listed in Table S1. PCRs were a relative estimation in triplicate as per the following temperature profile: denaturation 95 °C for 10 min followed by amplification by 40 cycles of 95 °C for 10 s and 60 °C for 1 min.
3. Total mRNA was isolated using Trizol (Invitrogen) and 2 μg RNA was used to synthesize cDNA using the PrimeScript RTreagent kit (Takara, DRR037A) according to the manufacturer’s protocol. Real-time PCR was performed using FastStart Universal Probe Master Mix (Roche) and analyzed with a Stratagene Mx 3000P thermal cycler. Real-Time PCR primer sequences are listed in Supplementary information, Table S1.
4. Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions, reverse transcribed and subjected to real-time PCR as previously described. [XX] Expression data were normalized to the geometric mean of the housekeeping gene GAPDH to control the variability in expression levels and calculated as 2- [(CT of indicated genes) – (CT of GAPDH)], where CT represents the threshold cycle for each transcript.
5. The total miRNA samples were extracted from ASPC-1 cells, PANC-1 cells and tumor tissues using SanPrep Column microRNA Mini-Preps Kit (Sangon Biological Engineering Technology & Services Co., Ltd., China) following the manufacturer’s protocol. The purity of the extracted miRNA was determined, and the RT-PCR was carried out using a microRNA First Strand cDNA Synthesis Kit following the manufacturer’s instructions with a TC-512 PCR system (TECHNE, UK). Quantitative real-time PCR assay was performed with a MicroRNAs Quantitation PCR Kit (Sangon Biological Engineering Tech- nology & Services Co., Ltd., China) and ABI 7500 Real Time PCR System (Applied Biosystems, USA). For the data of each sample, the Ct value of the target gene was normalized to that of U6 (Sangon Biological Engineering Technology & Services Co., Ltd., China). The unknown template was calculated using the standard curve for quantitative analysis. Eventually, the expression level of each gene was performed and the data were normalized to control unwanted sources of variation.
6. The total RNA of the tissue samples was extracted using the Trizol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA was converted from total RNA by using a Reverse Transcription Kit (Takara, Dalian, China) according to the manufacturer’s instructions. Quantitative real-time PCR was performed with SYBR Green (Takara, Dalian, China),0 and the data collection was performed on the Applied Biosystems® 7500 Real-Time PCR Systems (Thermo Fisher Scientific) according to the manufacturer’s instructions. The primers were synthesized by Biosune (Shanghai, China). The relative expression level of indicated genes was compared with that of β-actin and expression fold changes were calculated using 2-△△Ct methods. Each qRT-PCR reaction was performed in triplicate. Sequences of primers used for qRT-PCR in this study are shown in Additional file 1: Table S2.
7. Total RNA was extracted from cell lines, using Trizol total RNA isolation reagent (Invitrogen), according to the manufacturer’s specifications and treated with Turbo DNase (Ambion). cDNA was synthesized from total RNA (0.5 mg) using random hexamers with TaqMan cDNA Reverse Transcription Kit (Applied Biosystems). Gene primers (Gabra3 F-5’- GACCACGCCCAACAAGCT-3’; R-5’-AGCATGAATTGTTAACCTCATTGTATAGA-3’; GAPDH-F-5’-GAAGGTGAAGGTCGGAGTCAAC; R-5’-CAGAGT- TAAAAGCAGCCCTGGT-3’) were designed using Primer Express v3.0 Software and real-time PCR was performed using SYBR Select Master Mix (Applied Bio- systems). All reactions were carried out on the 7500 Fast Real-Time PCR System (Applied Biosystem). The average of three independent analyses for each gene and sample was calculated using the DD threshold cycle (Ct) method and was normalized to the endogenous reference control gene GAPDH.
8. TRIzol reagent (Thermo Fisher Scientific) was used to extract RNA from samples. Then a total of 250 ng of RNA was used to perform reverse transcription with QuantiMir RT kit (System Biosciences, Palo Alto, CA, USA). The primers used were as follows: CISD2, 5’-GCAAGGTAGCCAAGAAGTGC-3’ (forward) and 5’-CCCAGTCCCTGAAAGCATTA-3’ (reverse); GAPDH, 5’-GCGAGATCGCACTCATCATCT-3’ (forward) and 5’-TCAGTGGTGGACCTGACC-3’ (reverse). CISD2 was amplified as follows: step 1: denaturation at 95°C for 10 min; step 2: 40 cycles of 95°C for 15 s and 60°C for 1 min. Results were calculated with 2−DDCT and normalized to GAPDH.
9. Total RNA was extracted from cultured MCF7, A549 and UACC903 cells using Trizol reagent (Takara, Shiga, Japan) according to the manufacturer’s instruction. Reverse transcription was carried out with 0.5 μg total RNA using the PrimerScript™ RT reagent kit (Takara, Shiga, Japan). After incubation for 1 h at 42 °C, the reactions were terminated by heating at 70 °C for 15 min. To analyze alternative splicing of exon 2 in the Bcl-x gene, 5′ primer to Bcl-x (5′-GAGGCAGGCGACGAGTTTGAA-3′) and 3′ primer (5′-TGGGAGGGTAGAGTGGATGGT-3′) were used for PCR amplification (30 cycles, 94 °C, 30s; 56 °C, 30s; 72 °C, 1 min) with 2xEasy Taq superMix (Transgen Biotech, China). The length of splicing variants of Bcl-xL and Bcl-xS are 460 bp and 271 bp respectively. To analyze alternative splicing of exon 3, 4, 5, 6 in the Caspase 9 gene, 5′ primer to Caspase 9 (5′-GCTCTTCCTTTGTTCATCTCC-3′) and 3′ primer (5′-CATCTGGCTCGGGGTTACTGC-3′) were used for PCR amplification (30 cycles, 94 °C, 30s; 54 °C, 30s; 72 °C, 1 min). The length of splicing variants of Caspase 9a and Caspase 9b are 742 bp and 292 bp, respectively. PCR products were separated and analyzed on agarose gels, with the bands of the Bcl-x and Caspase 9 splicing variants being confirmed by DNA sequencing.
10. Brain tissues from the ipsilateral SN were dissected at the indicated time points after LPS or PBS injection with capsaicin or vehicle, and total RNA was extracted in a single step using RNAzol B (Tel-Test, Friendswood, TX) following the instructions of the manufacturer. Total RNA was reverse transcribed into cDNA using AMV reverse transcriptase (Promega, Madison, WI) and random primers (Promega). The primer sequences used in this study were as follows: 5′-TGATGTTCCCATTAGACAGC-3′ (forward) and 5′-GAGGTGCTGATGTACCAG TT-3′ (reverse) for IL-1β (378 bp); 5′-ACACTCTATCACTGGCATCC-3′ (forward) and 5′-AAGGGACACCCTTTCACAT-3′ (reverse) for COX-2; 5′-GCAGAA TGTGACCATCATGG-3′ (forward) and 5′-ACAACCTTGGTGTTGAAGGC-3′ (reverse) for iNOS (557 bp); and 5′-TCCCTCAAGATTGTCAGCAA-3′ (forward) and 5′-AGATCCACAACGGATACATT-3′ (reverse) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 308 bp). The PCR amplification consisted of 30 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s (for IL-1β and GAPDH or 54 °C for 30 s for iNOS) and extension at 72 °C for 90 s. PCR products were separated by electrophoresis on 1.5% agarose gels, after which the gels were stained with ethidium bromide and photographed. For semiquantitative analyses, the photographs were scanned using a Computer Imaging Device and the accompanying software (Fujifilm, Tokyo, Japan).


Этот информативный текст отличается привлекательным содержанием и актуальными данными. Мы предлагаем читателям взглянуть на привычные вещи под новым углом, предоставляя интересный и доступный материал. Получите удовольствие от чтения и расширьте кругозор!
Изучить вопрос глубже – https://vyvod-iz-zapoya-1.ru/
buy cytotec online – diltiazem brand purchase diltiazem online cheap
desloratadine 5mg for sale – loratadine pill cost dapoxetine 90mg
Thank you for this great piece of content. Best Regards
sildenafil 50mg usa – price viagra buy cialis 10mg
I don’t even know how I ended up here, but I thought this post was good. I don’t know who you are but certainly you’re going to a famous 🙂 Cheers!
tadalafil online order – sildenafil 100mg buy generic viagra 100mg
When I originally commented I clicked the “Notify me when new comments are added” checkbox and now each time a comment is added I get three emails with the same comment. Is there any way you can remove people from that service? Thanks a lot!
Undeniably consider that which you stated. Your favorite justification seemed to be on the internet the easiest thing to take into accout of. I say to you, I certainly get irked even as folks consider issues that they plainly do not realize about. You controlled to hit the nail upon the top as smartly as defined out the whole thing with no need side-effects, other people can take a signal. Will probably be back to get more. Thank you
Hello my loved one! I wish to say that this post is amazing, great written and come with almost all significant infos. I’d like to see extra posts like this.
noolthettam.com
buy clavulanate paypal – buy duloxetine generic duloxetine pills
Cheers, A lot of tips!
agen judi casino igkbet online https://eseomail.com/online-casino-video-poker/ win big 21 online casino
order crixivan generic – purchase crixivan sale buy voltaren gel
гѓ—гѓ¬гѓ‰гѓ‹гѓі йЈІгЃїж–№ – гѓ—гѓ¬гѓ‰гѓ‹гѓі гЃ©гЃ“гЃ§иІ·гЃ€г‚‹ г‚ёг‚№гѓгѓћгѓѓг‚Ї и–¬е±ЂгЃ§иІ·гЃ€г‚‹
バイアグラの飲み方と効果 – г‚·г‚ўгѓЄг‚№ её‚иІ© гЃЉгЃ™гЃ™г‚Ѓ タダラフィルジェネリック йЂљиІ©
order crotamiton online cheap – buy crotamiton cream purchase aczone gel
Thanks! I value this.
hard rock tampa online casino casino canada online slotomania online casino
Incredible many of amazing info!
online casino western union canadian online casinos bally’s twin river online casino
Many thanks, I like this.
mobile online casino eu top rated online casinos situs live casino online
Thank you. Numerous forum posts.
gta online diamond casino heist scope out casino online casino gta online casino heist avi schwartzman
Many thanks, Very good information.
online casino games az casino free online best casino online canada zodiac
You actually said that really well!
free online casino slot game spin casino canada bei welchem online casino kann man richtig geld gewinnen
Cheers. Quite a lot of information!
deutsche online casinos mit startguthaben realistic online casino double u casino online
Whoa all kinds of beneficial tips!
bandar judi casino baccarat online free online casino slot machines beste online casino zonder registratie
Factor well used!!
mohegan sun online casino login top online casinos canada casino slot online 888
Perfectly voiced without a doubt! .
online casinos ca real cash online casino magic wins online casino
You definitely made your point!
fanduel online casino pennsylvania best online casino sites canada ruby slots online casino
order flagyl online cheap – cenforce order buy cenforce 100mg pills
brand betnovate – betamethasone order online order monobenzone
brand acticin – how to get acticin without a prescription buy tretinoin
He looks down. And then smiles at his dad. His father unbuttons his shirt and throws it to the bathroom floor. Where his son is like the mythical David, cast in stone with blonde locks and cherubic face. The father is dark and with a day’s growth of stubble on his face.
“I thought you usually showered after practice. In the locker room.” His dad asks as he walks into the open door of the bathroom. “Once you plant that cock of yours in some squirmy hole and empty those warm balls of yours into a moist wet hole. Then you are on your way to becoming a man, and only then will you, come-of-age. But it is only a step onto the winding pathway towards manhood. It is my job to teach you what it means to be a man.” His father stands as he finishes his sentence. Loosening his belt and pulling his shirt out from the tucked confines of his pants.
Garrett does not go hog-wild on his tool. He caresses and adores it like one would an idol. porno dla dzieci Daddy was furious as I could hear his deep breath. He sat down on the bench by the door. I was looking down afraid he was going to kick my ass. He lifted my head from my chin using the tip of his loafer where I can smell that smelly sheer socks he has been worn all day. Just when our eyes met, a big stinky manly wad landed on my face. Then I felt a big rough hand rubbing the spit all over my face. I heard the daddy say, “ next time remember what I told you exactly, I don’t want this to happen again, you hear me? I paused and said, “ yeeee…”. Before I finished the sentence, another spit and a big slap on me. “ when I told you something you must say yes. There is no room for you to argue or think, understood?” He said in a deep and firm tone. “ yes sir”. Without missing a beep I said it. “good boy, your night is just about to start.”
Seriously a lot of excellent advice!
gta 5 online casino heist fingerprint [url=https://luckyusaplay.com/#]best paying online casino[/url] australian online mobile casino
You revealed it wonderfully.
online casino game free casino free online parx casino online betting
Great write ups. Regards!
oriental casino online online casino for real money usa free online no deposit casino
Thanks a lot! Numerous data.
mgm pennsylvania online casino online casino list treasure mile casino online
Really many of very good facts.
australia best online casino free casino games online casino online uk free
Thanks. A lot of material.
caesars casino ontario online online casino bonus no welches online casino
Lovely information, With thanks!
illinois online casino no deposit bonus online casino list all online casinos list
This is nicely said! !
online casino debit card deposit casino free games online online casino vulkan vegas
Seriously a good deal of great data.
new casino in michigan online best online casino welcome bonus no deposit ruby red casino online
You actually explained it well.
gta 5 online casino guide online casino no deposit bonus codes online casino blog
Thank you, I appreciate this!
news online casino top online casino blackjack casino online game
You stated that exceptionally well.
rival usa online casinos casino games free online 888b online casino
cheap trihexyphenidyl pill – trihexyphenidyl cheap voltaren gel online purchase
Many thanks, I value it.
christchurch casino online login casino bonus online video poker casino online
Seriously loads of wonderful tips!
usa online casinos free play play casino games online askgamblers casinosonline casinos 2019 best online casino sites askgamblers
You said it adequately..
9club casino online online gambling casino how to get approved for casino credit online
You made your position extremely effectively.!
no deposit promo codes for online casino online live casino casino royale movie online
Thank you! Awesome stuff.
michigan online casino new no deposit casino bonus usa online casinos firekeepers online casino and sportsbook
You revealed this very well.
online casino 5 dollar minimum deposit best online casino usa lucky master online casino
Good write ups. Thanks!
parx free online casino new online casino no deposit bonus usa nj online casino free money no deposit
With thanks. Wonderful stuff.
what’s the best online casino app online casino bonus free online casino with no deposit bonus
Nicely put, With thanks!
parhaat online casino bet online casino casinos gratis online tragamonedas
This is nicely put. .
casino online zdarma online casino sign up bonus online sweeps casino real money
You revealed that exceptionally well.
meadows online casino casinos online usa online casino lastschrift bezahlen
Very well expressed really! !
us-friendly online casinos us online casino online casino free credit no deposit
Cheers. Valuable stuff!
play online casino no deposit best casino online foxwood casino online
You said this exceptionally well!
begas casino online online us casinos casinos online for real money
Position well regarded.!
online casino fruit no deposit bonus online casino new online casino pa real money
Appreciate it! Ample postings!
bonanza slot online casino live casino online are online casinos mafoa
Nicely put, Appreciate it.
casino online luzern online casino for real money usa gta online casino vehicles
Kudos, I enjoy this!
online casino boobs free play online casino online casino bookie franchise reviews
You suggested that really well.
jugar online casino [url=https://luckyusaplay.com/#]live casino online[/url] online casino new york state
Cheers, A good amount of posts.
ph online casino online casino free bonus seriösen online casinos
With thanks! Numerous facts.
online casino booming games casino online casino deposit online casino
Thanks a lot! An abundance of facts.
dealer casino online new online casino usa ok top 10 online casinos in south africa
priligy dapoxetine 30mg Both tumor genomic factors 3 as well as heritable genetic factors may affect interindividual responses to drug therapy
order mobic 7.5mg generic – mobic oral buy toradol sale
Helpful tips. Thanks a lot!
best payout online casino uk https://usaplayerscasino.com/safe-casinos/ best online casino in bangladesh
buy baclofen tablets – feldene 20mg canada piroxicam 20mg generic
order diclofenac online cheap – order imdur 40mg sale order nimodipine online
Great info. With thanks!
me online casino bonus https://usacasinomaster.com/superbowl-betting/ best online casino real money no deposit free play
You said it perfectly.!
22bet online casino review https://luckyusaplay.com/credit-cards/ best american online casino
order generic pyridostigmine – buy generic azathioprine online generic imuran 25mg
Many thanks, Valuable information.
https://usagamblinghub.com/nascar-betting/
Excellent facts. Cheers.
https://uscasinoguides.com/real-money-bingo/
rumalaya online order – buy generic shallaki over the counter amitriptyline 50mg sale
buy cambia without a prescription – cheap aspirin 75mg generic aspirin 75 mg
buy besifloxacin online – purchase besifloxacin for sale order sildamax sale
calcort order online – brimonidine medication brimonidine online buy
0lom2r
order imusporin for sale – purchase imusporin sale order colchicine generic
buy generic lactulose – where can i buy mentat betahistine 16 mg usa
trileptal 300mg cheap – cheap pirfenidone cheap levoxyl tablets
oral terazosin – hytrin cost dapoxetine 60mg canada
buy cheap finax – alfuzosin 10 mg brand buy alfuzosin 10 mg sale
buy lasuna pills – how to get diarex without a prescription cheap himcolin tablets
buy gasex for sale – buy diabecon tablets cheap diabecon
Therefore, antimicrobial susceptibility testing should be performed on clinically significant C cialis online no prescription The smooth silicone has a dramatic curve at the tip to focus the deep, thrumming vibrations right where you need them
buy generic atorvastatin – buy bystolic without a prescription oral nebivolol 5mg
calan online order – diltiazem 180mg sale cheap tenoretic sale
tenormin tablet – oral carvedilol 6.25mg carvedilol medication
leflunomide 20mg ca – brand alfacip how to get cartidin without a prescription
rogaine where to buy – finasteride over the counter buy finasteride 1mg generic
cost zofran – detrol pill requip for sale
buy flexeril generic – buy olanzapine medication buy enalapril pills
cheap cytoxan generic – purchase cytoxan vastarel pill
generic divalproex – buy lariam generic topamax 100mg cost
hydrea pills – how to buy trecator sc buy robaxin tablets
piracetam 800mg usa – levofloxacin sale order sinemet 20mg sale
order piroxicam 20mg online – exelon 3mg brand exelon without prescription
how to get etodolac without a prescription – order pletal 100 mg generic order pletal without prescription
dimenhydrinate over the counter – cheap dramamine purchase actonel for sale
order fulvicin 250 mg online cheap – order generic gemfibrozil 300mg order lopid 300 mg generic
dapagliflozin pills – forxiga 10mg pills acarbose price
zovirax canada – eukroma canada buy duphaston 10 mg pills
bactrim 960mg generic – order bactrim 960mg online buy tobra 10mg generic
buy aciphex 20mg – generic maxolon generic motilium
cost bisacodyl 5 mg – order loperamide 2 mg for sale cost liv52
fludrocortisone distract – nexium pirate lansoprazole spread
clarithromycin rule – biaxin rent cytotec nearest
ascorbic acid risk – ascorbic acid greet ascorbic acid stoop
promethazine cloud – promethazine story promethazine sensation
priligy hell – dapoxetine anything priligy belief
loratadine rustle – claritin pills shimmer claritin pills score
loratadine medication pipe – claritin pills puzzle claritin pills total
valacyclovir couple – valtrex pills state valtrex monk
prostatitis pills universal – pills for treat prostatitis mention prostatitis medications love
uti antibiotics goose – uti antibiotics notice uti treatment bolt
asthma medication organ – inhalers for asthma or asthma medication certain
acne medication stre – acne medication basil acne treatment everything
cenforce online new – daily cialis brand viagra pills slip
cialis soft tabs union – tadarise online accept viagra oral jelly oxford
cialis soft tabs pills floor – viagra oral jelly online load viagra oral jelly corpse
brand cialis look – tadora fall penisole waste
cenforce cigar – cenforce light brand viagra online acquaint
priligy hunt – viagra plus hop cialis with dapoxetine however
viagra professional bottle – levitra oral jelly dozen levitra oral jelly online last
simvastatin air – fenofibrate police atorvastatin 40mg uk
crestor pills beam – crestor pills ultimate caduet home
nitroglycerin ca – cost combipres generic diovan 160mg
metoprolol online order – olmesartan medication purchase nifedipine
order microzide 25 mg generic – felodipine 5mg ca order generic zebeta 5mg
purchase digoxin generic – buy verapamil 240mg generic buy lasix sale diuretic
famvir online – valcivir 1000mg oral valaciclovir 1000mg usa
purchase ketoconazole for sale – oral itraconazole 100 mg order itraconazole sale
rybelsus price – desmopressin ca DDAVP usa
order lamisil 250mg for sale – griseofulvin cost griseofulvin online order
glyburide usa – generic glyburide 5mg brand dapagliflozin
methylprednisolone generic – order generic zyrtec astelin 10ml brand
clarinex 5mg ca – order clarinex 5mg pills buy allergy pills onlin
albuterol inhalator price – advair diskus inhalator price theo-24 Cr cost
buy ivermectin 12 mg online – eryc 250mg for sale order cefaclor 500mg online
zithromax brand – purchase ciprofloxacin generic buy ciplox generic
amoxicillin uk – buy cefadroxil 500mg buy cipro 500mg online cheap
augmentin pill – buy ciprofloxacin generic ciprofloxacin 1000mg cost
hydroxyzine 25mg canada – atarax 25mg price purchase endep
purchase seroquel without prescription – order sertraline 50mg pills buy cheap generic eskalith
clozapine 50mg drug – buy generic frumil over the counter buy pepcid 20mg generic
zidovudine 300mg without prescription – where to buy roxithromycin without a prescription buy generic allopurinol over the counter
order glucophage 1000mg sale – order generic glucophage 500mg order lincocin 500 mg online cheap
buy lasix without a prescription – order coumadin 2mg for sale purchase capoten pill
purchase ampicillin pill penicillin buy online buy amoxicillin no prescription
cheap metronidazole 200mg – cefaclor 500mg canada zithromax for sale online
EShqsbJWZydiaV
ivermectin 6mg pills – cefixime 100mg uk sumycin 500mg pill
buy valtrex 500mg online cheap – purchase diltiazem zovirax brand
ciplox online order – ciplox without prescription purchase erythromycin generic
cost metronidazole – cefaclor 250mg pills purchase zithromax generic
yPGntHMDfZXU
cipro drug – cephalexin 500mg generic buy augmentin 625mg generic
baycip where to buy – cephalexin 250mg generic amoxiclav pills
buy atorvastatin 80mg without prescription generic lipitor 80mg order atorvastatin 80mg pills
WAlzeQht