GROMACS的运行
GROMACS的运行过程可以分成以下三个阶段[1]:
(1) 前处理过程:生成模拟对象的坐标文件、拓扑结构文件以及平衡参数及其外力作用参数等文件。
(2) 模拟过程:首先要对系统进行能量最小化,避免结构的不合理而在模拟中出现错误;然后是对系统升温过程,先给系统的各个原子以Boltzmen分布初速度,再模拟较短的时间以达到初步的平衡;最后进行真正的分子动力学模拟,即平衡过程。此过程一般时间步长为1fs,运行时间在ns量级,以保证模拟系统尽可能找到势能的最低点。
(3) 后处理过程:MD模拟结束后,GROMACS会产生一系列文件,如.pdo文件(受力分析文件) 、.trr文件(模拟过程结果文件)、.edr文件(能量文件)等。同时,GROMACS本身还提供了多种分析程序,可以对这些文件进行分析,可以得到分子体系的各种信息。
安装教程
-
Windows 安装,请参考如下链接:
GROMACS的原生Windows版的编译和安装方法(支持GPU加速)
注: Windows 版安装时可选CPU版,GPU版加速版。电脑显卡水平较高的可选择GPU 加速版。
-
Linux 安装,请参考如下链接:
注:GROMACS通常在Linux下运行,此文章对其在Linux下的安装方法进行详细说明。另外当GROMACS版本的安装方法出现较大改变时,作者也会做相应的更新。
验证是否安装
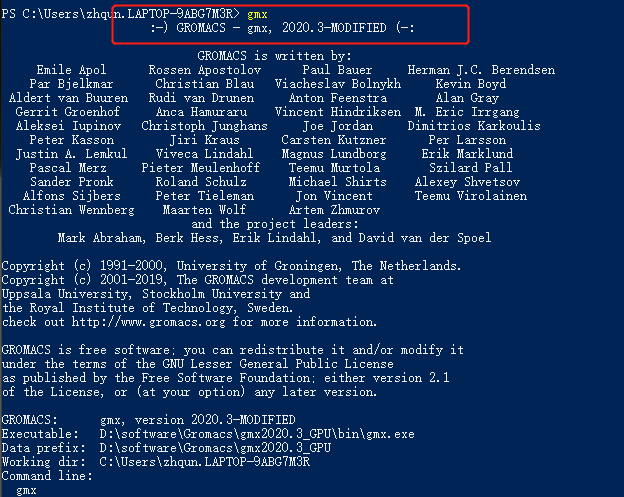
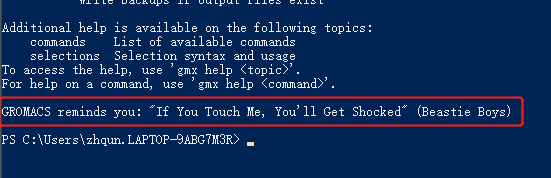
打开终端输入gmx 回车,如果出现的是一些gromacs软件版本介绍信息以及最后一句是两个井号括起来的名人名言就证明安装成功了。
如windows 系统中打开 WIndows Poweshell ,键入gmx,如果安装成功,会出现如下信息:
gromacs的版本号,以及在最后的一句名人名言。
GROMACS学习资料[2,3]
(1) GROMACS官网:
官网上不但含有软件下载资源,还有gromacs英文版说明书,内容比较全,英文好的同学可以尝试读一下。
(2) GROMACS说明书中文翻译:
此链接提供了gromacs英文说明书和英文步骤的中文翻译版,对于新手理解gromacs 的流程比较友好,建议仔细阅读。但希望看完中文的最好看一下英文的。
(3) 小木虫论坛:
小木虫论坛有分子动力学专栏,如果有什么问题,可以进行发布到论坛上,共同讨论解决。
(4) 知乎:
知乎上面有很多gromacs 的教程,搜索 gromacs 就有很多操作笔记以及运行参数调整等等的帖子,适合用于补充经验。
(5)bilibili:
B 站上有很多gromacs 的视频,介绍比较详细。看视频比纯看文字版说明书更容易学。最近在看一个up 主的视频:分子模拟从入门到发文章——1.gromacs的安装
感兴趣的可以学习吧。
(6)学习大牛的经验
a. sobereva
卢天是sobereva网名主要使用者, 从2006年左右开始长年累月通过计算化学相关论坛、QQ等方式义务解答我国青年计算化学工作者在计算研究中遇到的不计其数的问题(平均每年解答超过一万条问题),已帮助无数研究者解决了计算技术以及研究思想方面的问题。于2014年10月创办了计算化学公社(bbs.keinsci.com),是世界范围人气最高、讨论档次最高的计算化学论坛。
http://bbs.keinsci.com/forum.php
b. Jerkwin(李继存)
李继存老师写了大量的博文分析分子模拟中的经验、技巧、教程、程序等等,对分子模拟方面的学习和应用有着非常大的帮助。老师讲的很详细,极其建议阅读。
-
博文汇总
-
老师分享的程序,有一些gmxtool, 对gromacs的使用更加友好。
c. Justin A. Lemkul
GROMACS的大牛,写了GROMACS的几大教程,是GROMACS入门的不二选择。具体的教程英文原版在下面,网上也有很多翻译版本。该教程是英文版的,可能阅读起来有些吃力,但内容是相当正宗了,值得一学。
http://www.mdtutorials.com/gmx/index.html




digoxin 250 mg cheap – buy labetalol order lasix without prescription
order famciclovir 500mg generic – where can i buy acyclovir buy valcivir 1000mg generic
nizoral 200 mg drug – buy generic lotrisone sporanox online order
buy cheap generic semaglutide – buy desmopressin spray buy desmopressin generic
purchase terbinafine – generic fulvicin buy cheap griseofulvin
prandin ca – order prandin 2mg generic jardiance 25mg usa
cost micronase 2.5mg – purchase forxiga pills pill dapagliflozin 10mg
clarinex 5mg without prescription – buy clarinex generic buy ventolin paypal
oral albuterol – advair diskus inhalator order buy theophylline 400mg generic
ivermectin 3mg for sale – aczone online generic cefaclor 500mg
buy generic cleocin – terramycin 250mg ca buy generic chloromycetin
purchase amoxicillin pills – buy generic amoxicillin online ciprofloxacin 500mg drug
order augmentin 625mg sale – oral amoxiclav ciprofloxacin 500mg over the counter
hydroxyzine over the counter – buy endep 25mg online amitriptyline cost
cost clozapine 50mg – famotidine 40mg over the counter cost pepcid
generic retrovir 300mg – biaxsig sale zyloprim usa
order metformin 500mg generic – cost bactrim 480mg lincomycin usa
furosemide 100mg cheap – capoten 120mg generic buy capoten online
buy ampicillin cheap buy penicillin antibiotic order amoxicillin online cheap
cost metronidazole 200mg – buy amoxil generic zithromax 250mg drug
ivermectina online – buy generic ceftin online tetracycline sale
order valtrex 500mg pill – buy nemasole generic acyclovir 400mg generic
buy ciprofloxacin 500 mg online – order erythromycin 250mg generic erythromycin 250mg over the counter
order flagyl 200mg sale – azithromycin oral buy generic zithromax online
cheap ciprofloxacin – purchase baycip pill purchase augmentin
how to get baycip without a prescription – order keflex 250mg sale augmentin 625mg usa
order atorvastatin generic generic atorvastatin 40mg buy atorvastatin 10mg sale