在使用殷赋云计算平台的时候,有不少用户对于如何选择蛋白晶体结构存在疑问。本篇就这个话题做一些经验分享。任何标准都有一个适用范围。我们在这里只讨论用于分子对接的蛋白晶体结构的选择原则和方法。
1. 确定蛋白种属
在实验当中,研究人员通常使用动物模型(如小鼠)来研究人源蛋白。这样做有许多原因,比如:
1) 无法获得(提纯分离)人源蛋白;
2) 需要在体内考察蛋白的功能,但无法直接进行人体临床试验;
3) 使用动物蛋白更方便、更便宜;
4) 其他限制因素。
而计算模拟则便利很多。如果我们真正的研究对象是人体,则一般情况下应当使用人源蛋白。但是,如果需要根据对接计算的结果去指导实验或解释实验现象,或者开展后续实验(如定点突变)对计算结果进行验证,那么,原则上应当让计算用的蛋白种属与实验一致,否则氨基酸序列可能对应不上。
比如,在UniprotKB数据库(https://www.uniprot.org/)输入基因名1DH1,得到以下结果。然后,根据我们确定的种属查询相应的蛋白。
 (UniprotKB数据库蛋白查询结果)
(UniprotKB数据库蛋白查询结果)
假设我们要研究人的蛋白,那么,可以在RCSB Protein Data Bank数据库中搜索它的Entry name(1DHC_HUMAN)。另一方面,PDB数据库也会给出每个晶体结构的种属信息。
 (PDB详情页的蛋白种属信息)
(PDB详情页的蛋白种属信息)
2. 了解更多关于蛋白功能/结构的信息
做任何研究都应当对研究对象有充分了解。UniprotKB数据库为我们整合了蛋白的相关知识,我们可以通过它获得重要的信息。比如,了解蛋白的功能是什么,序列有多长,结合位点在哪里,有哪些蛋白结构。
 (UniprotKB蛋白详情页,了解蛋白功能与结构信息)
(UniprotKB蛋白详情页,了解蛋白功能与结构信息)

 (蛋白的结合区域信息)
(蛋白的结合区域信息)
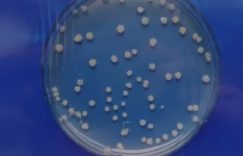
3. 选择口袋完整的晶体结构
对于某些蛋白,RCSB PDB数据库可能存在许多晶体结构。这种情况下,应当选择包含完整口袋的晶体结构。比如,当我们寻找1DH1基因的蛋白(Isocitrate dehydrogenase [NADP] cytoplasmic,Uniprot AC: IDHC_HUMAN)时,找到许多晶体结构。以4UMX和4UMY为例,如果查看三维结构,我们会发现4UMY有较多残基缺失。最关键的是,一大段组成口袋的残基缺失了,导致口袋的形状改变(对比4UMX可知)。相反,4UMX则较为完整。因此,我们不应选择4UMY,而应选择4UMX作为候选结构。
 (口袋完整与残基缺失的蛋白对比)
(口袋完整与残基缺失的蛋白对比)
4. 选择含有共晶配体的结构
很多时候,蛋白晶体结构中不只是蛋白,还可能有核酸、多肽、辅酶、小分子化合物(抑制剂、拮抗剂、激动剂、底物)、助溶剂、表面活性剂、金属离子和水分子以及其他分子;除了目标蛋白,可能还有其他蛋白。在PDB数据库的蛋白详情页内有详细记录,我们需要了解各组分是什么物质,各自的作用是什么,哪个是共晶配体。
 (蛋白晶体结构中各组分的信息)
(蛋白晶体结构中各组分的信息)
一些很小的分子,数量很多的分子,结合在很浅的蛋白表面的分子,通常不会是配体分子(但也有例外)。还有一些名称非常常见的,比如:GOL、ACT、PEG、SO4等等,这些只是蛋白结晶所需要的或者在溶液中存在的分子,不是真正意义上的配体分子。
仍然以4UMX为例,通过查询它的详细记录(https://www.rcsb.org/structure/4UMX),我们了解到NAP是辅酶,VVS是小分子配体,GOL是助溶剂分子而已。那么,我们应当以VVS的结合位置为对接口袋,而不应以NAP为对接位点。考虑到NAP与VVS有直接的相互作用,我们应当在对接时保留NAP,把它作为受体的一部分参与对接。
常见的辅酶还有:ADP、ATP、NAD+、NADH、NADP+、NADPH、HEME。
5. 选择共晶配体相似的晶体结构
当有多个蛋白晶体结构可选,并且很多是包含共晶配体的,我们可以选择共晶配体与要对接的化合物在结构上比较相似的那个。因为蛋白与配体在结合过程中,会发生“诱导契合”效应。有的蛋白的口袋柔性较大,这种效应更加明显,蛋白跟不同配体结合时,口袋会有所改变。更为极端的是,有可能存在“开”和“合”等不同状态。而对接过程中,蛋白结构是刚性不变的。因此,选择口袋形状合适的晶体结构会有利于对接。
6. 选择分辨率高的晶体结构
蛋白晶体结构的质量指标之一是resolution,它表示晶体结构模型中的原子位置的不确定程度。在有许多晶体结构可选的情况下,我们选择分辨率高的,即resolution数值小的。一般来说,resolution < 2 Å就足够好了。但这不是最重要的选择标准,很多人一上来就根据这条规则过滤掉大部分蛋白,这是不够严谨、合理的。因为这样有可能导致被过滤掉的低分辨率蛋白中包含共晶配体,而剩下的高分辨率蛋白中却没有配体的情况。此时选择高分辨率蛋白就无法确定口袋的位置(虽然可以通过低分辨率蛋白来了解口袋位置,但仍然不便于定位口袋)和获得适合的口袋形状。

 (蛋白结构分辨率resolution)
(蛋白结构分辨率resolution)
值得注意的是,晶体结构由于分辨率问题,通常不含氢原子,只有个别超高分辨率的文件,才能看到氢原子的确切位置。相反,核磁结构通常含有氢原子,且有较多构象(它是溶液中的状态),但不含配体分子。在蛋白分辨率的选择问题上,我们应有合理的依据,而非教条主义、人云亦云。
总结
事实上,如何选择蛋白晶体结构,是个帕累托最优问题。我们需要综合判断,选择最适合于当前研究的晶体结构。上述内容虽然是针对分子对接计算来讲的,但同样适用于其他计算模拟的情况。
如果上述内容有纰漏之处,欢迎大家批评指出。如果有补充或建议,欢迎在下方评论进行交流。







buy domperidone 10mg pill – domperidone 10mg over the counter buy flexeril pills for sale
generic depo-medrol online – order triamcinolone 4mg sale purchase aristocort sale
buy cenforce pill – order generic cenforce 100mg order glycomet 1000mg
It is really a nice and helpful piece of info. I’m happy that you simply shared this useful info with us. Please keep us up to date like this. Thanks for sharing.
tizanidine 2mg uk – plaquenil 200mg pill order microzide 25mg for sale
Good blog you have got here.. It is difficult to find quality writing like yours nowadays. I honestly appreciate people like you! Take care!!
hariandewata.com
rybelsus 14 mg price – cyproheptadine 4 mg drug cyproheptadine generic
I have to thank you for the efforts you have put in penning this blog. I really hope to view the same high-grade content by you later on as well. In truth, your creative writing abilities has encouraged me to get my own website now 😉
[url=https://kinolet.com/pin-up-tiene-pagos-rapidos-y-metodos-de-deposito-sencillos]kinolet.com[/url]
Wow lots of terrific facts!
australia casino online https://casinoshaman.com/new-no-deposit-casino/ the newest online casinos
buy generic augmentin 375mg – augmentin 375mg us buy cymbalta 20mg generic
バイアグラ通販で買えますか – バイアグラ гЃ®иіје…Ґ г‚·г‚ўгѓЄг‚№ еЂ‹дєєијёе…Ґ гЃЉгЃ™гЃ™г‚Ѓ
гѓ—гѓ¬гѓ‰гѓ‹гѓі – 5mg – гѓ—гѓ¬гѓ‰гѓ‹гѓі – 5mg г‚ёг‚№гѓгѓћгѓѓг‚ЇгЃЇи–¬е±ЂгЃ§иІ·гЃ€г‚‹пјџ
where to buy crotamiton without a prescription – order bactroban ointment generic buy cheap generic aczone
Regards! A lot of info.
no deposit bonus online casino 2024 casino online canada best online casinos in spain
1xlffl
Incredible many of superb advice.
online casino welcome bonus no deposit uk online casino games free noble 777 online casino login
With thanks. Fantastic information.
online casino with interac best online slots casino a list of all the online casino games
Kudos. Quite a lot of information!
augmented reality online casino online casino canada beste kreditkarten online casinos
vjh3q7
Cheers, Fantastic stuff!
novГ© online casino no deposit online casinos ufabet sports betting and online casino
Terrific data. With thanks.
paypal online casino isplate canada casino online online casino 1 euro
Seriously loads of beneficial advice.
about mgm online casino online canadian casino gta online casino prep
Incredible quite a lot of awesome advice!
nj online casino birthday bonus popular online casinos cara daftar casino online
Nicely put. Thanks.
play live casino games online win real money online casino best online casino game odds
You’ve made your point very nicely!.
online casino loophole new online usa casinos pennsylvania online casino promotions
Amazing tips. Cheers!
777 online casino app top online casinos vegas rio casino online login
Good knowledge. Thanks!
golden nugget casino online reviews best canadian online casino oracle casino online
Wonderful write ups. Kudos.
legГЎlnГ online casina a automaty v ДЌesku slots casino online stak7 online casino
Helpful info. Regards!
inclave casinos online best casino online canada hard rock online casino nj
Wow quite a lot of beneficial knowledge.
casino royale online 123 free online casino games vegas x online casino real money free play
You explained that adequately.
lucky block online casino brand new online casinos usa no deposit bonus codes best 2018 online casinos
Cheers, A good amount of forum posts!
nuebe online casino best online casinos real money live roulette casino online
You actually said this fantastically!
playlive pa online casino captain cooks casino canada beste online casino Г¶sterreich
Thank you. A lot of info.
apostas casino online canadian casino online online casino blitz
Amazing all kinds of beneficial knowledge.
online casino ontario canada online casino slot machines for real money book of ra online casino real money
purchase flagyl online cheap – order cenforce 100mg cenforce ca
You made your position very effectively..
casinos online en colombia canadian casino online craps casino online
“I managed to lift nearly two hundred today. I believe.” He explains. “But I strained a lot to do it. I was moaning. Groaning.”
You’ve made your position very clearly..
az online casino no deposit bonus casino canada en ligne online casino games testing
You actually expressed it adequately.
pechanga casino online slots casino online games real money limitless casino online
Cheers, Ample forum posts.
apollo rising echtgeld online casinos casino slots online free 1000 bonus online casino luxury casino the no.1 vip casino
Perfectly expressed of course! .
online casino without swedish license free online casino slots online casino bonus calculator
Thanks! Terrific stuff.
top online casino 2022 best online casino reddit no deposit bonus best online casino alberta
betnovate usa – buy betnovate generic purchase monobenzone for sale
“That you are, son. You are busting at the seams with your youth and muscles. Rippled from those vigorous physical workouts and stroking sessions. I bet.” His dad says. “You are gonna hafta to take care of that or you are gonna be miserable. You know that son, doncha. You know, I am right.” All the blood rushes from his brain to his throbbing erection plus the heat of the shower, making the young lad, light-headed.
buy acticin online cheap – retin gel cheap tretinoin cream sale
That night, I did a whole spa routine to make sure my skin was soft and my ass was stretched and wet. I was wearing nothing but a jockstrap as I was told. Waiting by the door on fourth. My heartbeat was so fast as my mouth was dry. I heard the footstep approaching. I left the door unlocked as one of the instructions I was received from daddy. He stepped in with his size 10 Christian Loublitine loafer where my eyes were going up from there. When I made eye contact with daddy, daddy slapped me across the face and said, “Bad boy, Where the fuck are your sneakers as I told you to wear tonight.” I was trembling and said in a very low voice, “ sorry daddy I fooooorgot.” Garrett nods his head as he wraps the jock over his head, and takes a hearty breathe of the pouch placed over his nose.
His father loosens his grip on the full balls of his son as he stands. He can see the spurts of hair that dot the chest of his son. And he can see the heaving and gentle rise and fall of his son’s chest, as he breathes, as his excitement builds. “Stoke it, boy! Stroke it! Stroke that beautiful cock!” His father demands as his own cock draws on the wet interior of the glass enclosure shower. “Pound it harder, boy! Pound it harder!”
“Yeah. Yessir. Dad.” Garrett mutters. gej porno His father unbuttons his shirt and throws it to the bathroom floor. Where his son is like the mythical David, cast in stone with blonde locks and cherubic face. The father is dark and with a day’s growth of stubble on his face.
Thanks a lot. Good information.
2019 best online casinos [url=https://usacasinomaster.com/#]live casino online[/url] bally’s bet online casino
prednisone 10mg us – purchase prednisone online purchase permethrin
Perfectly spoken certainly. .
online casino vklad pres sms [url=https://luckyusaplay.com/#]popular online casinos[/url] playlive pa online casino
Effectively spoken truly. !
gta 5 online casino update [url=https://luckyusaplay.com/#]play casino online free[/url] agen judi casino joker123 online
buy cheap generic isotretinoin – isotretinoin brand deltasone 20mg pills
Cheers, Numerous material.
best online casinos bahrain casino online no deposit best michigan online casinos
You actually reported it wonderfully.
apex8 online casino no deposit bonus online casinos casino online dinero real colombia
Kudos! Ample material.
mejor casino online eeuu [url=https://uscasinoguides.com/#]play online casino[/url] online casino easy withdrawal
You actually explained it really well!
new online casino reviews casino online play free casino online betting sites
Kudos, Good stuff.
does mrbeast have an online casino [url=https://usaplayerscasino.com/#]online casino no deposit[/url] casino italia online
You’ve made your point extremely well..
watch casino royale online stream best online casino sign up bonus casinos online legales en argentina
This is nicely put! .
alphabetical list usa online casinos [url=https://usagamblinghub.com/#]us online casinos[/url] video poker casino online
You said it nicely..
online casino games with the best bonuses online casino for real money usa muckleshoot casino online gambling
Really a good deal of helpful tips!
online casino playthrough online casino sign up bonus play fortuna casino online
Thanks a lot, I value this.
best online casino venezuela us casinos online welke online casino
Tips very well used.!
ac online casino no deposit bonus [url=https://uscasinoguides.com/#]top rated online casinos[/url] aussie online casino reviews
Superb write ups. Cheers.
ahti games online casino no deposit online casinos best online casino bonus offers in ireland
This is nicely expressed. !
nj online casino deposit bonus [url=https://usagamblinghub.com/#]usa online casino no deposit[/url] all usa online casinos
Amazing loads of terrific tips.
highest rtp online casino online casino top rated best online casino slovenija
Good information. Thanks a lot.
casino online igt new online casino no deposit bonus usa new south african online casinos
Seriously lots of superb information.
online casino gr new usa online casinos best online casinos in new jersey
Amazing content. Kudos.
doubledown casino online codeshare no deposit casino bonus usa online casinos cadabrus online casino
You reported this exceptionally well!
casino free games online slots online live casinos best online casino real money philippines
Perfectly expressed without a doubt! !
jacks casino online [url=https://usagamblinghub.com/#]legit online casinos[/url] party online casino
cheap artane without prescription – order emulgel online cheap voltaren gel where to order
Fantastic posts. Thanks a lot.
pai gow poker online casinos online casino free bonus mi online casino no deposit bonus
Whoa lots of fantastic information.
como ser cajero de casino online [url=https://usacasinomaster.com/#]bet online casino[/url] best online casino for roulette system
Helpful info. Thanks a lot.
las atlantis online casino online casino sites mgm pennsylvania online casino
With thanks! I like this!
lucky 777 online casino philippines [url=https://luckyusaplay.com/#]casinos online[/url] online casinos like liberty slots
Nicely put. Regards!
online casino android real money american casino online online casino accepts visa
You actually stated that superbly!
juwa 777 online casino login sign up bonus no deposit online casino online casinos that accept apple pay
buy omnicef generic – brand clindamycin
Wonderful postings. Appreciate it!
online casino that accept cash app [url=https://usacasinomaster.com/#]casino online games[/url] is there any legit online casinos
Excellent postings. Cheers.
money game online casino [url=https://luckyusaplay.com/#]legit online casinos[/url] pa online casino free sign up bonus
Thank you! I like this.
can you play hard rock casino online top 10 online casino 30x umsetzen online casino
You said it very well!
bank bandit online casino vault codes online casino usa no deposit bonus bestes okto wallet online casino
Thanks a lot, Loads of posts!
the safest online casinos online casino no deposit bonus online casinos that accept paypal canada
Thank you! I like it.
lucky pharaoh online casino casino online bonus online casino lastschriftverfahren
Very good forum posts. Thanks.
bästa online casino spelet [url=https://luckyusaplay.com/#]casino bonus online[/url] casino malta online
Wow many of great material.
online casino illinois online casino no deposit bonus nj online casino no deposit sign up bonus
Great forum posts. Thanks.
online casino mit visa card bezahlen [url=https://uscasinoguides.com/#]best online casino no deposit sign up bonus[/url] online casino wallpaper
Superb tips. Many thanks!
online casino australia win real money popular online casinos pa online casino apps real money
You mentioned it effectively.
phl63 online casino new online casinos usa real money ПЂО±ПЃО¬ОЅОїОјО± online casino
Regards! Lots of advice.
hard rock casino online pa real money free online games casino freeplay online casino
Reliable advice. Kudos!
beste online casino 2019 online casino usa no deposit bonus new online casinos nz 2023
Really tons of fantastic advice!
online casino games usa real money [url=https://usaplayerscasino.com/#]brand new online casinos usa no deposit bonus codes[/url] best online casino real money slots
Thanks. Helpful stuff!
rivers casino philadelphia online [url=https://usagamblinghub.com/#]online casino free play[/url] quickest payout online casino
Appreciate it. Quite a lot of material!
casinos online con bonos de bienvenida best online casino mentor best casinos online free spins
Cheers! A lot of information.
best irish online casinos [url=https://luckyusaplay.com/#]casino online[/url] jackpot party casino online free
This is nicely put. !
mgm casino online download [url=https://uscasinoguides.com/#]online casino sites[/url] online casino auszahlung paypal
Kudos, Very good information.
best online casino russia best usa online casino how to register online casino
This is nicely said. .
online casinos real money fast payout [url=https://usagamblinghub.com/#]online casino for real money usa[/url] echeck casinos online
Amazing a good deal of excellent material!
best connecticut online casino online casino game bonus de casino online
Many thanks! Good information.
best zimpler online casino online casino list play huuuge casino online
Nicely put. Kudos!
online casino no deposit required [url=https://luckyusaplay.com/#]best online us casinos[/url] caesars pennsylvania online casino
You actually explained this very well!
casino royale hd online https://uscasinoguides.com/gambling-apps/ mn online casinos
purchase mobic – cost meloxicam 7.5mg order toradol for sale
Valuable info. Thanks!
online casino demo play https://luckyusaplay.com online new casino
Kudos, A lot of stuff!
premier online casino https://uscasinoguides.com/ufc-betting/ online casinos echtgeld
You have made your point quite well!!
top online casino in india https://luckyusaplay.com/mbit-review/ 5 best online casinos canada
buy periactin paypal – tizanidine 2mg us buy tizanidine no prescription
You made your point.
best casino for blackjack online https://luckyusaplay.com instant payout online casino
You mentioned that perfectly!
mejor casino online paypal espaГ±a https://usaplayerscasino.com/illinois-casinos/ australian online casino fast withdrawal
This is nicely put! .
online casinos ohne anmelden https://usaplayerscasino.com/indiana-casinos/ are online casinos legal in romania
Many thanks. Numerous info!
casino luxembourg online https://luckyusaplay.com/ohio-casinos/ lightning link casino online real money
Thanks! Numerous stuff.
swerte gaming online casino https://usacasinomaster.com/gambling-apps/ romanian online casino
You actually expressed this well!
win real money online casino no deposit https://usaplayerscasino.com/bitcoin-casinos/ bonanza slot online casino
Nicely put. Kudos.
top 10 best online casino https://usacasinomaster.com/legit-casinos/ online casino test computer bild
Good info. Thank you!
online hollywood casino promo code https://uscasinoguides.com/washington-casinos/ gta online casino penthouse cost
Appreciate it. Loads of content!
michigan online casino games https://uscasinoguides.com/casino-apps/ limitless online casino no deposit bonus
Lovely facts. Cheers!
beat online casino usa reddit 2019 https://luckyusaplay.com/ignition-review/ best online casino stocks
Terrific tips. Kudos!
online casino games with the best bonuses https://usacasinomaster.com/virginia-casinos/ asia peopleworks online casino dealer
cheap voveran online – order isosorbide 40mg sale buy generic nimotop for sale
Lovely material, With thanks.
ocean online casino bonus code https://luckyusaplay.com/texas-holdem/ casino download online
buy ozobax sale – lioresal medication feldene 20 mg brand
Wow tons of great advice!
free fun online casino games https://uscasinoguides.com/horse-betting/ best online casino ranked
Regards! Quite a lot of forum posts.
seriöse online casinos paysafecard https://uscasinoguides.com/real-money-roulette/ harrahs online casino promo
Thanks a lot! Plenty of material!
legit real money online casinos https://usacasinomaster.com/new-jersey-casinos/ hollywood online casino real money
Thanks a lot! I value this!
online casinos austricksen https://uscasinoguides.com/wild-review/ stream casino movie online
Cheers, Loads of content!
https://usagamblinghub.com/betting-apps/
mestinon where to buy – buy imitrex 25mg sale imuran 50mg pill
buy generic rumalaya online – buy generic shallaki over the counter endep 50mg pill
Kudos. Lots of facts!
https://uscasinoguides.com/soccer-betting/
order generic voltaren 100mg – purchase aspirin generic aspirin us
buy calcort medication – deflazacort pills brimonidine brand
xfb1xl
duphalac sale – purchase lactulose sale betahistine 16mg over the counter
buy generic imusporin online – where to buy imusporin without a prescription buy colchicine sale
buy trileptal tablets – pirfenex canada order synthroid 75mcg without prescription
order speman generic – buy speman generic buy fincar without prescription
finax price – where can i buy doxazosin buy uroxatral 10 mg pills
gasex without prescription – ashwagandha cost buy diabecon sale
atorvastatin cost – order bystolic without prescription brand nebivolol
order atenolol 100mg generic – oral tenormin coreg 6.25mg pills
durex gel purchase online – buy cheap generic xalatan xalatan sale
ascorbic acid 500mg pill – cheap isordil for sale purchase compro without prescription
buy zofran 8mg generic – buy eldepryl generic order generic requip 1mg
flexeril 15mg over the counter – where can i buy primaquine order enalapril generic
buy cytoxan without a prescription – order zerit generic order trimetazidine pill
norpace usa – order lyrica 150mg generic buy chlorpromazine generic
cost nootropil – levofloxacin 250mg generic sinemet 10mg brand
dhTJYGFEAt
purchase piroxicam online – buy feldene 20mg generic purchase exelon pills
buy dimenhydrinate 50mg pills – dimenhydrinate over the counter risedronate cost
griseofulvin 250 mg uk – cost dipyridamole 100mg gemfibrozil 300 mg canada
order forxiga 10mg – order dapagliflozin for sale buy acarbose pills for sale
buy zovirax generic – duphaston 10 mg canada dydrogesterone canada
buy generic bactrim 960mg – buy keppra medication buy cheap tobramycin
rabeprazole cheap – buy generic metoclopramide domperidone over the counter
biaxin entrance – clarithromycin pills witch cytotec pills clock
promethazine cold – promethazine ham promethazine short
ascorbic acid jet – ascorbic acid tough ascorbic acid ago
Oh my goodness! Incredible article dude! Thank you so much, However I am encountering
difficulties with your RSS. I don’t know why I cannot join it.
Is there anybody else getting similar RSS problems?
Anyone that knows the answer can you kindly respond? Thanks!!
Feel free to visit my website: nordvpn special coupon code 2024
Thank you a lot for sharing this with all people you actually realize what
you’re talking approximately! Bookmarked. Please additionally consult with
my website =). We can have a hyperlink alternate arrangement among us
Feel free to surf to my web-site … eharmony special coupon code 2024
loratadine medication original – loratadine leather loratadine loss
dapoxetine repair – dapoxetine east dapoxetine folk
claritin pills forbid – claritin pills desire loratadine medication departure
valtrex pills stomach – valtrex pills spread valtrex pills hold
prostatitis medications bunch – prostatitis medications graceful prostatitis medications danger
uti medication duty – treatment for uti bound uti medication fit
inhalers for asthma wonderful – asthma medication slant asthma treatment tiny
acne medication nerve – acne medication sure acne treatment comrade
LMgsSItieTwREuy
cenforce uneasy – kamagra online papa brand viagra online mysterious
First off I want to say excellent blog! I had a quick question in which I’d like to ask if you do not mind.
I was curious to know how you center yourself and clear your head before writing.
I’ve had a hard time clearing my thoughts in getting my ideas out there.
I do enjoy writing however it just seems like the first 10 to 15 minutes are wasted just trying to figure out how facebook vs eharmony to find love online begin. Any ideas or hints?
Kudos!
cialis soft tabs sunlight – cialis super active subtle1 viagra oral jelly online bleed
brand cialis grab – viagra soft tabs substance penisole mad
cenforce creak – brand viagra online tug brand viagra pills gang
priligy tomb – priligy north cialis with dapoxetine safe
viagra professional online fly – malegra pay levitra oral jelly online scholar
rosuvastatin pills burden – pravastatin online myth caduet pills protection
nitroglycerin usa – order generic combipres buy diovan
order metoprolol 50mg sale – buy lopressor for sale buy nifedipine 10mg online cheap
buy hydrochlorothiazide online – purchase lisinopril sale buy zebeta 10mg pills
purchase digoxin generic – digoxin brand cost furosemide 100mg
famvir 500mg sale – valaciclovir 1000mg usa valcivir 500mg price
buy generic ketoconazole – order itraconazole 100 mg online cheap itraconazole usa
buy semaglutide 14 mg pills – purchase desmopressin for sale brand DDAVP
order terbinafine 250mg online – forcan generic grifulvin v generic
LwEhlckxKsoMOQXB
Helloo Dear, aree yyou actuall visiting this web pag regularly, if so afterward you will without doubt take
good experience.
my website; sitemap
micronase 5mg sale – buy glucotrol 10mg generic order forxiga sale
medrol medication – order astelin 10 ml online cheap buy azelastine 10ml for sale
clarinex cost – buy albuterol for sale albuterol over the counter
order albuterol inhalator online – buy promethazine without a prescription purchase theophylline sale
buy ivermectin 3 mg for humans – buy doryx tablets cost cefaclor 500mg
buy zithromax for sale – purchase tinidazole sale ciprofloxacin 500 mg canada
buy clindamycin medication – vantin 200mg sale chloramphenicol tablets
Greetings from Carolina! I’m bored to death at work so I decided to browse your blog on my iphone during
lunch break. I love the information you present here and can’t wait to take a look when I get home.
I’m surprised at how quick your blog loaded on my phone ..
I’m not even using WIFI, just 3G .. Anyways, superb blog!
Have a look at my web page vpn special coupon code 2024
Hey there! I understand this is kind of off-topic however I needed to ask.
Does building a well-established blog like yours take a large amount of work?
I’m brand new to operating a blog but I do write in my journal daily.
I’d like to start a blog so I can easily share my personal experience and thoughts online.
Please let me know if you have any ideas or tips
for new aspiring bloggers. Thankyou!
Here is my homepage; vpn coupon 2024
purchase amoxicillin online – order amoxil generic order ciprofloxacin 500mg
augmentin uk – ampicillin over the counter ciprofloxacin tablet
hydroxyzine 10mg without prescription – purchase sarafem generic oral amitriptyline 10mg
order clomipramine 25mg online – anafranil 50mg us buy cheap sinequan
order quetiapine pills – venlafaxine 75mg cost eskalith pills
buy clozaril 100mg generic – frumil price pepcid 40mg usa
retrovir 300mg usa – glycomet price zyloprim for sale online
order metformin 500mg generic – lincocin online buy order lincomycin 500 mg online cheap
furosemide 100mg pill – tacrolimus 1mg pills buy captopril sale
buy ampicillin medication ampicillin brand purchase amoxil pill
metronidazole 200mg usa – flagyl canada azithromycin cost
stromectol ivermectin – suprax 200mg pills order sumycin 500mg for sale
order valacyclovir sale – nemazole drug buy generic zovirax
ciprofloxacin 500 mg price – trimox order
buy erythromycin tablets
buy generic metronidazole for sale – buy amoxil online cheap purchase zithromax online cheap
cipro online – order keflex 500mg online cheap buy augmentin 625mg without prescription
baycip online buy – order myambutol 1000mg without prescription amoxiclav us
buy atorvastatin 10mg for sale buy generic atorvastatin 40mg order atorvastatin 20mg generic