但凡接触过DNA、蛋白序列的人,或多或少都用过BLAST。作为应用最广的生物信息学线上工具,它的操作界面十分简洁。给使用者的直接印象就是选择你要的功能—递交你的序列—点击提交得到结果,但背后的原理确实十分复杂。BioEngX小编就带你多了解一些BLAST的背后,希望能让各位用BLAST更加得心应手。
BLAST的发展史
BLAST最早是由NCBI(The National Center of Biotechnology Information)在1990年提出并引入使用的。那个时候的BLAST功能十分有限,只能无缝对比未知序列和已知功能序列,给出的统计学数据只能引导出两序列是相似还是不相似这一结论。直到1997年,有缝对比的算法才被引入,也就是我们今天看到的BLAST的基础模型。随后,每年都会引进几种新功能,比如后来的PSI-BLAST,不断地扩充形成了今天BLAST的规模:多功能,多数据库对比。现在的BLAST不仅仅是把一条序列送去和已知序列数据库对比这么简单,同时还具备分析序列所属物种分类,分析功能区域等同步功能。
BLAST的应用界面
如下图所示,这是现在的BLAST主页的样子(https://blast.ncbi.nlm.nih.gov)
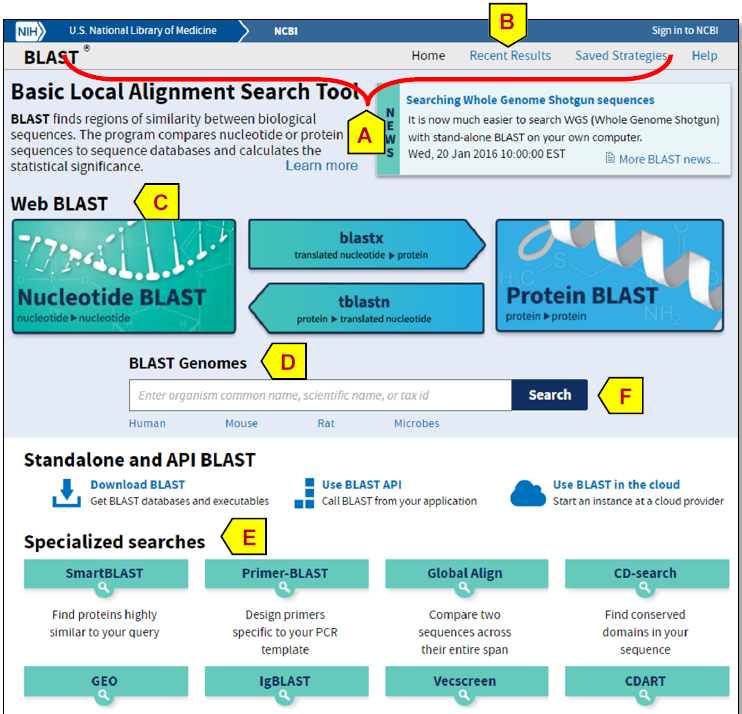
A部分为BLAST主页的抬头
B部分为查询最近搜索结果
C部分就是BLAST的主要部分了,分别显示了四种主要的BLAST方式,Nucleotide BLAST ,Protein BLAST,blastx,tblastn,这是根据输入数据的类型和数据库类型来定义的。
D和F 部分就是限定在固定的物种里面进行BLAST
E部分就是更加具体细化的BLAST功能组件
不同的序列不同的BLAST方法
BLAST的方式有很多,选择哪一种主要取决于你的输入序列的类型和你想要发现的相关信息,如果你知道自己想找什么类型的结果,那用起BLAST就会方便很多。在这里简单介绍几个最常用的BLAST功能。
BLASTN
用核酸序列去对比核酸序列数据库,主要用于鉴别未知序列所属的物种类型和了解未知序列在其他物种基因组的分布情况。
BLASTP
用蛋白序列去对比蛋白数据库,寻找同源蛋白,鉴别蛋白功能区间,确定蛋白的分类等等功能。
BLASTx
输入核酸序列,对比蛋白数据库。一段未定义过的未知核酸序列的翻译方式一共有六种,5’>3’ frame 1、2、3,和3’>5’ frame 1、2、3。BLASTx就是将核酸翻译成这六段,然后通过对比蛋白数据库寻找核酸上可能存在的基因序列的位置。
tBLASTn
就是反向BLASTx,通过对蛋白序列进行所有可能的逆转录得到各种可能的核酸序列,再去对比核酸数据库,用以确定蛋白所属物种和具体基因的位置。
PSI-BLAST
Position-Specific-Iteration BLAST,定点循环蛋白BLAST方法,其实就是先进行基础的BLASTP,找到相关的类似蛋白,然后我们在所有的相似结果中选择一批蛋白,程序会帮我们总结出这些相似蛋白的共同氨基酸点位,并根据这些点位来搜寻其他相似蛋白。这个方法极大提高了可搜寻同源蛋白的范围,能比较全面的知道你的输入蛋白和哪些蛋白具有相似位点。
除了这些以为,BLAST还有更多细化的对比方式,就按具体情况具体使用咯。
Reference
Altschul SF et. al. Basic local alignment search tool. J Mol Biol. 1990 Oct 5;215(3):403-10.
管理员邮箱:info@bioengx.org;管理员微信:bioengxadmin; 内容和图片来自网络。欢迎留言讨论哦,如需再转载,请联系管理员。
扫描下方二维码关注BioEngX官方微信公众平台









ketoconazole 200mg generic – how to buy lotrisone buy itraconazole 100 mg pill
famciclovir 250mg over the counter – buy valaciclovir generic valaciclovir 500mg ca
terbinafine for sale – griseofulvin pills buy grifulvin v pills
Wow, marvelous blog structure! How long have you been blogging for?
you made running a blog glance easy. The full look of your site is great, as well as the
content material! You can see similar here sklep online
methylprednisolone 4 mg pills – oral singulair 5mg buy azelastine generic
stromectol coronavirus – eryc online buy cefaclor 250mg sale
order cleocin – cost vantin 200mg chloramphenicol drug
zithromax us – tindamax for sale order ciprofloxacin 500 mg generic
purchase amoxil online cheap – cephalexin 250mg drug order generic cipro 1000mg
order augmentin 375mg sale – buy cheap generic ethambutol buy ciprofloxacin no prescription
cheap hydroxyzine 10mg – how to get endep without a prescription endep without prescription
clomipramine 25mg ca – anafranil 50mg uk order sinequan 25mg
retrovir sale – epivir 100mg canada brand zyloprim 100mg
clozaril online order – buy amaryl 1mg online cheap pepcid 20mg brand
order glycomet 1000mg generic – buy cheap lincomycin purchase lincomycin online cheap
buy lasix online cheap diuretic – buy generic prograf over the counter captopril 25mg drug
purchase flagyl – order metronidazole 200mg buy azithromycin 250mg generic
acillin tablet order vibra-tabs without prescription purchase amoxil pills
purchase valacyclovir generic – valacyclovir 500mg pills how to buy acyclovir
can i buy ivermectin online – order ciplox 500mg generic generic sumycin
metronidazole 200mg sale – order flagyl 400mg online cheap order azithromycin 250mg for sale
buy generic ciprofloxacin online – erythromycin 250mg cheap erythromycin buy online
cipro 1000mg cheap – keflex 250mg uk order augmentin 375mg
buy cipro 500mg without prescription – purchase ethambutol generic augmentin canada
lipitor price atorvastatin online buy atorvastatin 10mg sale