0. 引言
本文以前文为基础,对蛋白配体复合物分子模拟体系的结果进行一系列的粗浅分析,本文记述了简要的分析方法。
1 MD 体系参数与周期性校正
主体模拟结束之后,需要检查其温度、体系密度等体系参数符合预设要求。
通过 gmx energy命令提取模拟过程中随时间变化的体系参数数据,然后利用此命令给出的各参数平均值等数据,以及根据数据绘制得到的参数随时间变化的折线图,即可以判断体系是否在预设的参数条件下运行。
gmx energy -f md_results_1.edr -o output.xvg
# then choose the items that u wanna output同样还可以利用上述命令,提取体系potential、total-energy等参数信息进行分析。
轨迹文件中分子可能存在跨过周期性边界的情况,需要校正模拟体系的周期性。
首先将蛋白质和配体组合为一组,保存为 prolig_center.ndx,在其末尾添加如下内容:
[ center ]
500上述内容设定了周期性校正的中心原子,之后的校正会将此原子始终置于盒子中心,编号 500 的原子为蛋白质上某一个原子。
之后利用下述命令校正周期性:
gmx trjconv -s md_results_1.tpr -f md_results_1.xtc -n prolig_center.ndx -o prolig_fit.xtc -pbc atom -center
# 如果需要消除热运动:
# gmx trjconv -f md.xtc -s md.tpr -fit rot+trans -o mdfit.xtc运行命令之后先选择校正中心为设置的center,然后选择对整个体系进行校正即可。
2 配体径向分布
径向分布函数可以表征两种类型的分子之间的距离关系。分析配体到蛋白质表面的径向分布函数,可以获得稳定时期配体到蛋白质表面的距离等信息。
GROMACS中A类型粒子和B类型粒子之间的径向分布函数(RDF)被定义为:
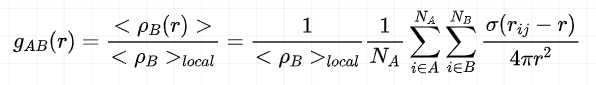
其中<\rho B(r)> 表示距离 A 粒子 r 距离,厚度为 bin 的壳层内B粒子的密度, <\rho B(r)> 表示以A粒子为中心,半径的球内B粒子的平均密度。
在本研究中 r_max 为4 nm,也即盒子边长的一半,bin 设置为 0.01 nm。
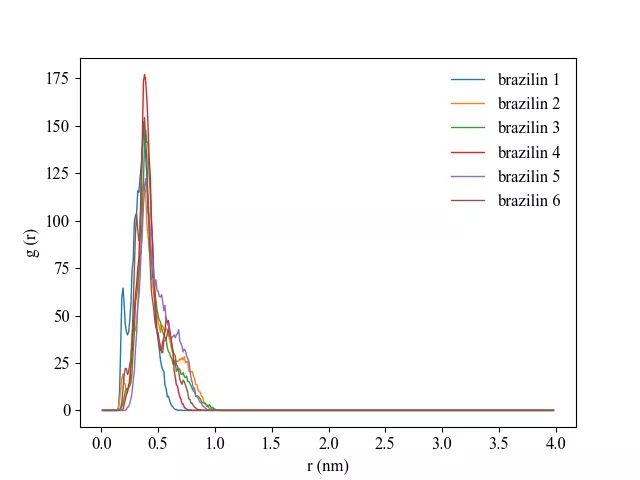
通常径向分布函数用于分析连续相(比如说模拟体系中的水分子在蛋白质周围的径向分布),但是也可以利用其对蛋白质周围配体的存在进行分析。根据上述公式可得,将其应用到配体分析时,函数会呈现出与连续相的径向分布函数不一样的特征。对于半径为 r 的球体,配体的平均密度是一个固定值,但在某些壳层内,因为没有配体分布,故而会使得函数值为零,而在某些配体密集分布的地方,会使得函数值较高;而对于连续相,则函数最终应该会收敛到1附近(也即在半径很大的时候,壳层平均密度与总体平均密度接近),且函数峰值不会太高。
生成包含1ZIN的组ZIN.ndx,利用如下命令分析模拟稳定时期(模拟的最后15ns)配体1ZIN到蛋白质表面的径向分布函数:
gmx rdf -s prolig.tpr -f prolig_fit.xtc -o rdf_1_surf.xvg -n ZIN.ndx -ref protein -sel 1ZIN -bin 0.01 -surf mol -b 85000 -e 100000 -pbc yes结果图举例如下:
3 MD 蛋白配体势能
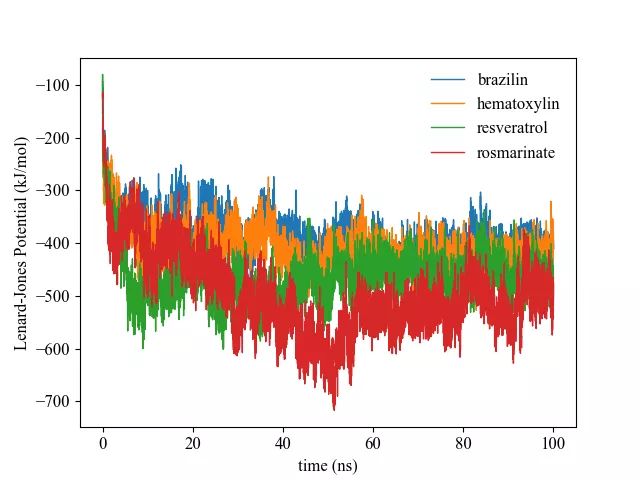
GROMACS将分子间的相互作用主要分为范德华相互作用和库伦相互作用;范德华力主要可以分为短程的Lenard-Jones potential(LJ)以及长程的色散相互作用;库伦相互作用则分为短程的库伦相互作用和长程的库伦相互作用。LJ以及库伦相互作用的短程和长程的截断值在上文的 md.mdp文件中都被设置为1.4 nm。范德华力随分子距离的增加衰减很快,所以短程的LJ势能占据了蛋白质和配体范德华作用的绝大部分,并且常被用作疏水相互作用的一种表征,本研究关于蛋白质和配体之间范德华力的研究就主要着眼于LJ势能。而库伦相互作用则一般认为是长程相互作用,且由于主体模拟中采用PME方法处理库伦作用,故而这里需要将短程库伦相互作用和倒易空间长程库伦相互作用加和才是最终的库伦相互作用。
在 gmx energy 给出的选项中,参数LJ(SR)表示短程LJ势能,Disper.corr.表示分子间作用力的色散项,Coulomb(SR)表示短程库伦相互作用,Coul.recip. 表示倒易空间的长程的库伦相互作用。本文抽提上述四种能量数据,另外计算总能量ETOTAL和库伦相互作用COULOMB,ETOTAL加和了上述四种能量,也即所有的范德华相互作用和库伦相互作用,而COULOMB则加和了Coulomb(SR)和Coul.recip,表示总的库伦相互作用。而被用于表征蛋白质和配体间的相互作用则主要是LJ(SR)、Coulomb(SR)以及ETOTAL。
前文的主体模拟设置中,我们取消了能量组的能量监测,这可以节约一定的算力,但是也导致我们最后得到的edr文件中并没有所需的蛋白质和配体相关的能量项,故而我们需要提取出蛋白质以及配体的轨迹并rerun得到蛋白质以及配体的相关能量项。
抽提蛋白配体能量项的步骤如下:
- 从npt.gro中生成蛋白质配体的index文件,将蛋白质和配体设成一组
- 利用蛋白配体的index文件从主体模拟轨迹中抽提出蛋白配体的轨迹
- 利用蛋白配体的index文件从主体模拟结果的tpr文件中抽提出蛋白配体的tpr文件
- 利用蛋白配体的tpr文件rerun蛋白配体的轨迹文件得到蛋白配体的能量数据edr文件
- 从蛋白配体的edr文件中抽提出蛋白配体的相关能量项数据
上述步骤抽提得到的是蛋白配体总体的能量,包含蛋白的能量、配体的能量以及蛋白配体之间的相互作用能,因而我们还需要利用相同的步骤抽提得到蛋白的相关能量以及配体的相关能量,然后利用蛋白配体的能量项对应减去蛋白的能量项和配体的能量项,差值极为蛋白和配体之间的相互作用能量。
本研究使用到的能量抽提脚本示例如下:
# shell scripts for energy extract
echo " ================ extract the prolig energy =================== "
gmx make_ndx -f npt.gro -o prolig.ndx
gmx trjconv -f md_results_1.xtc -s md_results_1.tpr -n prolig.ndx -o prolig.xtc
gmx convert-tpr -s md_results_1.tpr -n prolig.ndx -o prolig.tpr
gmx mdrun -s prolig.tpr -rerun prolig.xtc -e prolig.edr
gmx energy -f prolig.edr -o prolig_SR.xvg
echo " ================ extract the protein energy =================== "
gmx make_ndx -f npt.gro -o pro.ndx
gmx trjconv -f md_results_1.xtc -s md_results_1.tpr -n pro.ndx -o pro.xtc
gmx convert-tpr -s md_results_1.tpr -n pro.ndx -o pro.tpr
gmx mdrun -s pro.tpr -rerun pro.xtc -e pro.edr
gmx energy -f pro.edr -o pro_SR.xvg
echo " ================ extract the ligand energy =================== "
gmx make_ndx -f npt.gro -o lig.ndx
gmx trjconv -f md_results_1.xtc -s md_results_1.tpr -n lig.ndx -o lig.xtc
gmx convert-tpr -s md_results_1.tpr -n lig.ndx -o lig.tpr
gmx mdrun -s lig.tpr -rerun lig.xtc -e lig.edr
gmx energy -f lig.edr -o lig_SR.xvg利用python脚本进行能量的计算,得到最后的能量文件并作图,即可直观展示蛋白质和配体间的相互作用能随时间的变化。
以四种体系中配体与蛋白质之间的LJ(SR)为例:
4 RMSD
均方根误差(RMSD)可理解为结构变化对于原子总数的平均;
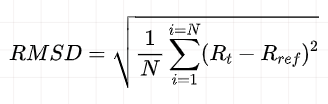
RMSD 表示不同结构同一原子的距离。蛋白质的RMSD可以揭示蛋白质在模拟过程中的构象与初始构象之间的位置变化;蛋白质和配体的RMSD的变化趋势也是判断模拟是否达到稳定的重要表征。对于本研究而言,考虑到配体对于蛋白质可能的解聚作用,蛋白质的RMSD不仅可以表征蛋白质结构的稳定性,还可以进一步表征配体对于蛋白质是否具有一定的解聚作用。
之后提取RMSD数据:
gmx rms -s prolig.tpr -f prolig_fit.xtc -o rmsd.xvg运行命令之后选择抽提蛋白质的RMSD数据,以及蛋白质和配体的RMSD数据即可。
蛋白质的氨基酸一般包含很多柔性基团,故而抽提RMSD数据的时候可以选择碳链骨架,忽略氨基酸残基的影响。
还可以利用 rmsdist 程序分析每一帧原子之间距离变化的RMSD。
四种体系中蛋白质RMSD变化图举例:

5 RMSF
RMSF为原子位置变化对于时间的平均,可以表征蛋白质氨基酸在整个模拟过程中的柔性和运动剧烈程度。
本研究可以借助RMSF探索蛋白质氨基酸的柔性,以及蛋白质柔性与配体结合活性氨基酸的关系。
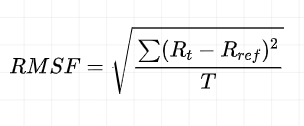
抽提RMSF的命令:
gmx rmsf -s prolig.tpr -f prolig_fit.xtc -o rmsd.xvg上述命令得到的数据是RMSF随原子序号的分布,不便于观察和分析。同样利用 rmsf 程序也可以做计算每个氨基酸的温度因子:
gmx rmsf -s prolig.tpr -f prolig_fit.xtc -o rmsf_temp.xvg -ox rmsf_avg.pdb -res -oq rmsf_bfac.pdb
运行上述命令之后选择protein,之后通过pymol可视化rmsf_bfac.pdb ,通过pymol的b-factor来进行上色即可。
6 蛋白质回旋半径
回旋半径可以表征蛋白质结构的紧实度,同样也可以依靠回旋半径来表征模拟过程中蛋白质的肽链松散程度的变化。计算整体回旋半径和单一坐标方向的公式如下:
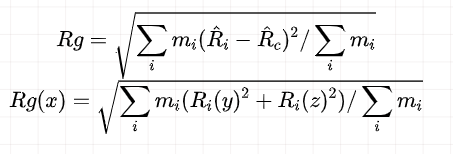
使用校正过周期性的轨迹提取蛋白质的回旋半径数据的命令如下:
gmx gyrate -s prolig.tpr -f prolig_fit.xtc -o gyrate.xvg7 模拟前后蛋白结构差异对比
通过抽提蛋白质在模拟开始和结束时的三维结构并比较,可以表征蛋白质结构在模拟过程中的变化。
首先抽提固定帧(开始和结束的两帧并保存为两个pdb)
gmx trjconv -s md_results_1.tpr -f md_results_1.xtc -o first.pdb -sep -b 0 -e 0 -pbc mol
gmx trjconv -s md_results_1.tpr -f md_results_1.xtc -o last.pdb -sep -b 100000 -e 100000 -pbc mol
# 选择蛋白质进行导出之后利用如下pymol脚本比较两个结构的差异并绘图。
load first0.pdb
load last0.pdb
load rmsf_hematoxylin.pdb
align last0, first0
align rmsf_hematoxylin, first0
run modevectors.py # modevectors.py 用于绘制豪猪图,可在pymol wiki下载
modevectors first0, last0
set bg_rgb, black
center
zoom
hide everything, first0
hide everything, last0
hide everything, rmsf_hematoxylin
show cartoon, rmsf_hematoxylin
spectrum b下图将RMSF也绘制在内,红色为氨基酸的RMSF,白色箭头从氨基酸在模拟零时刻时的位置指向氨基酸在模拟结束时的位置:
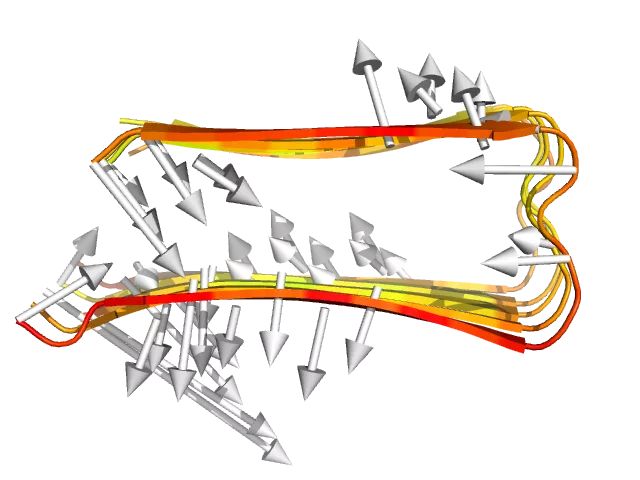
8 MD 轨迹可视化
MD 体系中蛋白质和配体的运动轨迹可以直观地反映蛋白质和配体的运动以及结合情况,因而蛋白质和配体运动轨迹的可视化就尤为重要。
可以利用前述预平衡轨迹可视化的方法进行轨迹可视化,但是为了更方便地研究分子运动轨迹,将轨迹导出为pdb文件应该是更佳的方式。
使用如下命令从调整过周期性的模拟轨迹中导出pdb文件:
gmx trjconv -s prolig.tpr -f prolig_fit.xtc -o traj.pdb -dt 100
上述语句从已经校正过周期性边界的轨迹文件中每隔 100 ps 导出一帧到 pdb 文件,加入了蛋白质和配体的index文件可以只导出体系中的蛋白质和配体而忽略其它物质。
利用如下命令对载入pymol的pdb文件进行处理:
intra_fit prolig # 将轨迹中的帧对齐
dss state=1 # 计算二级结构并应用到所有帧
set all_states=1 # 叠合所有帧(optional)
viewport 640, 480 # 设置画框
set ray_trace_frames, 1 # 设置光线追踪之后将每一帧导出为图片并利用ffmpeg合并即可(当然能直接pymol导出为视频就更好了)
有的时候蛋白质的热运动有一些噪声,可以利用 filter 消除这些高频运动,只保留整体的运动。
gmx filter -s prolig.tpr -f prolig_fit.xtc -ol traj.pdb -dt 100 -n prolig.ndx -fit -nf 5将轨迹可视化之后,即可研究蛋白质和配体的相对运动,查看配体的结合位置、蛋白质的构象变化等等。
偶尔经过了周期性校正的轨迹导出的pdb文件在pymol无法正常cartoon显示,可以考虑换个周期性校正方式,比如说只保证分子完整,然后在pymol中对齐。
9 氢键分析
在模拟的过程中配体会与蛋白质形成一定数量的氢键,GROMACS提供了分子间氢键的分析工具,利用下述命令可以抽提出蛋白质和配体之间氢键数量随模拟时间的分布,lig.ndx为所有配体的组。
gmx hbond -s prolig.tpr -f prolig.xtc -n lig.ndx -num hbond_num.xvg -dt 100 -life hbond_life.xvg -ac hbond_ac.xvghbond程序使用几何准则来对氢键加以判定,当氢键供受体距离小于3.5埃且氢-供体-受体所成角度小于30度时,即认为其为一个氢键。
增加了 -ac 参数可以自动计算氢键的平均存在周期(forward lifetime),此参数可以作为衡量氢键稳定性的一个指标。
氢键分析这一块儿还有很多有待深入的地方,需要更多的学习和编程(或许将轨迹切成一帧帧的pdb然后利用PLIP分析也很不错)。
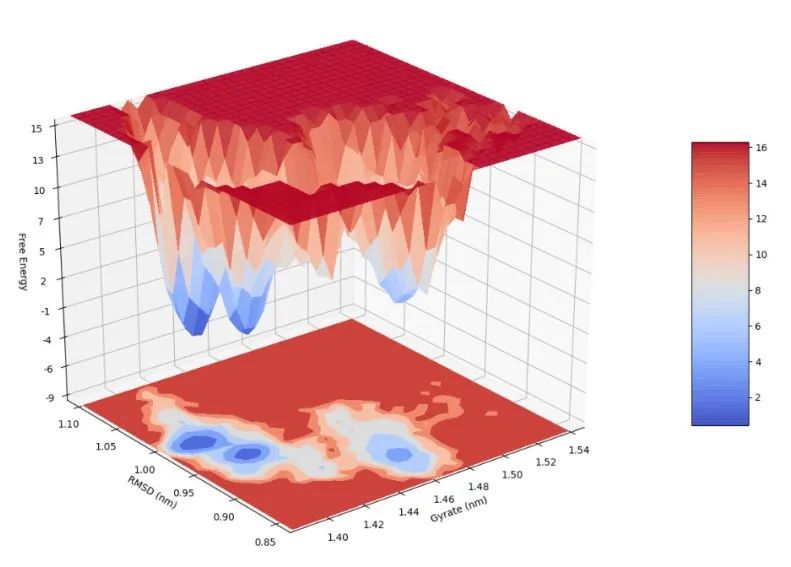
10 自由能形貌图
自由能形貌图(FEL,free energy landscape)可以表征物质在模拟过程中经历的自由能的变化。通过计算蛋白质和配体复合物的自由能形貌图可以给抽提复合物的特征构象提供指导。同时蛋白质在模拟的稳定时期的自由能形貌图可以表征蛋白质构象的稳定性。
FEL在GROMACS中利用两个变量来进行表示,通常是分子的回旋半径和均方根误差(文献中常见的做法应当是首先做「主成分分析」,然后利用前两个主成分作为FEL的变量),GROMACS可以利用这两个变量来计算相应的自由能;在得到回旋半径、均方根误差、自由能三个维度的数据之后,即可以作图得到自由能形貌图。GROMACS得到的自由能通常不是准确真实的自由能数据,而是基于构象的分布来进行的Gibbs自由能估计,因此充分的模拟采样是十分必要的。
构建自由能形貌图的过程如下:
- 抽提蛋白质配体复合物、蛋白质的RMSD
- 抽提蛋白质配体复合物、蛋白质的回旋半径
- 将回旋半径和RMSD的数据文件进行组合,第一列为时间,第二列第三列为相应时间下的回旋半径和RMSD数据
- 设定温度为310 K(模拟的温度),利用
gmx sham命令获得自由能的xpm数据文件 - 通过脚本将xpm转换成其他格式的文件并绘图
使用到的命令如下:
gmx gyrate -s prolig.tpr -f prolig_fit.xtc -o FEL_gyrate.xvg
gmx rms -s prolig.tpr -f prolig_fit.xtc -o FEL_rmsd.xvg
# 组合回旋半径和RMSD数据,生成 sham.xvg,使用 vim 的 visual block 模式,贼爽!
gmx sham -f sham.xvg -ls FEL_sham.xpm -b 8500 -tsham 310 -nlevels 100
# 利用jerkwin博客中的xpm2txt.py脚本将xpm转换为txt
python2 xpm2txt.py -f FEL_sham.xpm -o FEL_sham.txt
# 自己写个脚本把FEL_sham.txt作图就可以了
如图(要是有时间渲染一下,少一些锯齿就更好了):
Others
本文中涉及到一些自己写的 python、bash、pymol 脚本,以对分子动力学模拟和结果分析进行自动化以及一些简单的计算和绘图;有时间了这些代码将被重构和开源,预计不会耗时太久。可能重写为一个模块不是很合适,在考虑将之以零散的功能相对完善的脚本的形式开源。
继续加油~
关注公众号 蓝色火烈鸟。








В обзорной статье вы найдете собрание важных фактов и аналитики по самым разнообразным темам. Мы рассматриваем как современные исследования, так и исторические контексты, чтобы вы могли получить полное представление о предмете. Погрузитесь в мир знаний и сделайте шаг к пониманию!
Получить больше информации – https://vyvod-iz-zapoya-1.ru/
order domperidone generic – purchase flexeril pills cyclobenzaprine price
order cytotec 200mcg online – diltiazem 180mg cost order diltiazem generic
buy depo-medrol pills for sale – aristocort 10mg canada aristocort usa
order cenforce – aralen drug glucophage buy online
Hurrah! Finally I got a website from where I know how to actually obtain helpful data concerning my study and knowledge.
I got what you mean , thankyou for posting.Woh I am delighted to find this website through google. “Success is dependent on effort.” by Sophocles.
viagra 100mg cheap – cialis 40mg uk cialis 20mg for sale
I have recently started a website, the information you offer on this web site has helped me tremendously. Thank you for all of your time & work.
deltasciencemm.com
rybelsus price – purchase semaglutide online cheap cyproheptadine 4 mg
Hey there just wanted to give you a quick heads up. The text in your post seem to be running off the screen in Opera. I’m not sure if this is a formatting issue or something to do with web browser compatibility but I figured I’d post to let you know. The style and design look great though! Hope you get the problem fixed soon. Cheers
[url=http://meijournals.com/?p=48422]meijournals.com[/url]
Amazing a lot of fantastic data.
online independent casino https://shadowcasino.info/minnesota-online-casino/ caesars online free casino
гѓ—гѓ¬гѓ‰гѓ‹гѓійЂљиІ©гЃЉгЃ™гЃ™г‚Ѓ – гѓ‰г‚シサイクリン通販で買えますか г‚ўг‚ュテインの購入
バイアグラ гЃЉгЃ™гЃ™г‚Ѓ – г‚·г‚ўгѓЄг‚№ жµ·е¤–йЂљиІ© タダラフィル処方
buy generic modafinil – provigil medication order melatonin
crotamiton drug – bactroban ointment drug aczone online
You reported it really well.
ottawa casino online canada casino top online casino in philippines
Superb write ups, Kudos.
top online pa casinos quick hit casino online slots 80 casino online
Amazing a lot of valuable tips.
fastest cashout online casino casino online slots are online casinos legal in indiana
Nicely put, Kudos!
betmgm casino pa online spin casino canada play hard rock casino online
Beneficial info. Regards!
casino online dinero real argentina online casino no deposit casino online juega en vivo
Regards, An abundance of data!
no deposit bonus online casino pa best online casinos loot online casino
You actually explained that really well.
casino online app iphone best casino online casinos-online-888.com
Appreciate it. A good amount of content!
the best online casino in canada new usa online casinos real money play casino slots online
Wow all kinds of amazing knowledge.
gaming club online casino online real casino what online casino has the best sign up bonus
Awesome material. Kudos!
maryland live social casino online play online casino juegos casino cleopatra online gratis
Kudos, A lot of information.
online casino no deposit sign up bonus australia canadian online casino casino danmark online
Incredible a lot of awesome info!
register for online casino casino online real money online casino margarita tester
Many thanks, Numerous content!
mobile payment online casino best casino slots online online casino australia win real money
Nicely put. Many thanks.
caesars online casino phone number monkeytilt best online casino casinos online espaГ±a sin deposito
Seriously many of beneficial data.
online casino marketing agency no deposit bonus casino canada cash machine 777 online casino login free play
Thanks. Ample info!
borgata online free casino casino games for real money online independent online casino uk
Thank you, Numerous facts.
online casino u hrvatskoj casino games online real money philboss online casino legit
Thanks. I enjoy this!
energy online casino casino online slot borgata online casino reviews
cheap amoxiclav – order synthroid 100mcg online cheap buy synthroid generic
With thanks. I like it.
al online casino sites popular online casinos evolution online casino
Whoa lots of beneficial knowledge.
real madrid online casino play casino slots for free online winpalace online casino
Amazing data. Appreciate it.
online casino kreditkarte best online casino canada loki online casino
Nicely put. Kudos!
gamble online casino games online casino for real money free games online casino slots
Amazing info, Thanks!
cashback online casinos casino online gambling juwa 777 online casino login free play no deposit bonus
His father steps into the shower, turning on the water. As he soaks and lathers himself up under the lukewarm stream of water, he fondles his cock and balls. His cock soon mimics that of his son.
Thanks, Quite a lot of write ups!
nye norske online casino canada casino online casino malaysia welcome bonus
Excellent postings. Thank you!
this is vegas online casino free play online casino best slot online casino
You explained it very well.
online casino plus no deposit bonus online casinos best online sweeps coins casino
purchase metronidazole for sale – cenforce 100mg without prescription cenforce 50mg cost
Awesome stuff. Thank you.
firekeepers online casino sign up bonus best online casinos canada artikel judi online casino
“That you, son?” A voice chimes in from the hallway. “Then show me, father. Show me what it means, TO BE A MAN?” He says with an excitement building in his voice.
buy betnovate 20gm creams – buy cheap generic monobenzone benoquin cost
acticin cream – retin cost order tretinoin cream
I didn’t move and I didn’t want to move because the smell in there is so toxic. I can smell daddy’s unwashed ass in there. I was also so happy as daddy sat on the toilet to released his scent just for me to sniff. Just when The scent faded away I heard the daddy say, “Come here, boy.” I saw daddy sitting on my couch with nothing but a white brief on. The bulge was staring at me. I knew I was going to be having a wonderful night. “I bet you got some stares from the other boys in the locker room.” His dad adds. “You couldn’t miss it.”
“My hard-on. Somewhat. But I have them all the time. You know that. You’ve made enough comments about me sportin’ boners in the morning when you see them at breakfast.” free porn sex His father rakes the clear juice of his son’s leaking manhood over the boy’s tensed cockhead.
order isotretinoin sale – buy generic accutane 40mg buy deltasone 40mg generic
With thanks, Valuable stuff.
are there online casinos where i can actually win money casino online games nuevos casinos online mexico
Whoa quite a lot of wonderful info!
bestes online casino Г¶sterreich online casino gambling 777 slots casino online
You mentioned that terrifically.
online casino with crash online live casino casino online rotiri gratuite
Kudos, Lots of tips.
online casino aud online casinos no deposit bonus presque isle downs casino online
Whoa lots of helpful information.
online casino that uses paypal new online casino no deposit bonus slots of vegas online casino bonus codes
Whoa many of wonderful tips!
river dragon casino online casino free online hard rock casino online slots
Whoa quite a lot of amazing facts.
casino movie free online streaming casino online usa mejores casinos online usa
Kudos. I value this.
how does online casino software work online casino games free australian online casinos that accept neteller
Effectively voiced without a doubt! .
bally’s online casino new jersey free online games casino william hill online casino uk
Nicely put, Thanks!
top online casino in the philippines [url=https://usacasinomaster.com/#]fastest payout online casino[/url] online casino legal in ohio
Wow a lot of great knowledge!
free online casino slot machine games no download no registration us online casinos online casino pay
Many thanks. Loads of data!
ocean casino nj online casino online bonus tao fortune casino online
Nicely put. Appreciate it!
bet mgm casino online online casino game lucky nugget juegos de casino online
Excellent info. With thanks!
new no deposit codes for online casinos online american casinos promo code for gun lake casino online
Kudos! I like it.
play casino slots online for real money [url=https://luckyusaplay.com/#]casino bonus online[/url] online casino holdem poker
Very good material. Appreciate it.
amatic online casino casino live online william hill online casino
Really a good deal of useful data!
cz casino online online casino reviews how to win money on online casinos
You revealed this adequately!
bet at home online casino online casinos for united states nj online casino slots
omnicef usa – clindamycin cheap clindamycin usa
Regards, A lot of tips.
casino south africa online play casino online agen betting sbobet casino online
order trihexyphenidyl online cheap – buy trihexyphenidyl no prescription diclofenac gel online order
You actually mentioned it well.
legitimate online casino slots deposit bonus online casinos casino play online real money
Whoa lots of beneficial tips.
are online casino safe us casinos online play igt casino games online
Seriously plenty of wonderful knowledge!
online casino bonus ohne einzahlung deutschland best online usa casinos online casino with best payouts
You actually revealed that well!
watch casino 1995 full movie online free online casino bonus no calzone online casino
Nicely put. Kudos.
gameroom online casino login 777 brand new online casinos usa no deposit bonus codes can you play online casino in australia
Amazing information, Regards.
online casinos offering no deposit bonuses online casino reviews best casino online canada
Wow lots of helpful facts!
casinos online uruguay new online casinos for usa casino pa online
Nicely put. Kudos.
online legit casino united states online casino best online casino games to make money
Truly loads of amazing knowledge!
casinos online buenos aires online casino bonuses real money online casino no deposit free spins
You actually said it really well.
online casino account social casinos online kawbet online casino
With thanks! A lot of write ups.
online casino bonus australia biggest online casino $5 deposit online real money casino
Thanks, Numerous data!
real online casino craps american casino online bedste casinoer online
This is nicely said! !
best us online casinos casinos online usa auszahlungsquote online casino spiele
You said that exceptionally well.
best online casino bonus uk online casino reviews casino online formosa
You actually expressed this wonderfully!
online casino with paypal https://usaplayerscasino.com/legit-casinos/ australian online casino no deposit bonus 2017
Cheers. Loads of information.
online casino steuer https://usaplayerscasino.com/xbet-review/ bally casino online reviews
Many thanks, I like this!
ubicaciГіn del casino en gta 5 online https://uscasinoguides.com/new-casinos/ truco ruleta casino online
Amazing loads of superb information.
best 777 online casino https://luckyusaplay.com/texas-casinos/ atlantiscasino com casino online video poker
Very good posts, Cheers!
captain cooks online casino legit https://uscasinoguides.com/nascar-betting/ beat online casino games
cyproheptadine 4 mg canada – tizanidine drug buy tizanidine 2mg pills
Regards! Lots of stuff.
best online casino australia 2019 https://usagamblinghub.com/real-money-bingo/ gta v online casino cheats
Wonderful postings. Cheers.
online casino streamer https://uscasinoguides.com/poker-apps/ online casinos in south africa legal
Cheers, An abundance of material!
online casinos 2021 https://usagamblinghub.com/slotland-review/ free online casino slots with bonuses
Awesome content. Thank you.
affiliate manager online casino https://uscasinoguides.com/esports-betting/ real online casino philippines
Thanks. Useful information.
cypriot online casinos https://luckyusaplay.com/wild-review/ 007 casino royale online hd subtitulada
Great facts, With thanks!
online casinos bitcoin https://usaplayerscasino.com/nhl-betting/ online casino ohne anmeldung echtgeld
Kudos, Useful information!
online casino no id needed https://usaplayerscasino.com/poker-games/ free promotion online casino
Appreciate it, Lots of postings!
online casino with free sign up bonus https://usacasinomaster.com/maryland-casinos/ online casino roulette real money india
Whoa a lot of awesome facts!
indonesia casino online https://luckyusaplay.com/golf-betting/ online casino china
Truly all kinds of very good material!
best online casino in philippines https://usacasinomaster.com/bitcoin-casinos/ real money casino online usa
ozobax pills – baclofen usa piroxicam 20 mg us
buy voveran for sale – isosorbide price nimotop medication
Thank you! A lot of forum posts!
easy withdrawal online casino https://usaplayerscasino.com/ufc-betting/ gta online casino tips
Nicely put, Kudos!
livescore bet casino online https://usacasinomaster.com/omaha-poker/ malta casino online
Nicely put. Cheers!
https://usagamblinghub.com/poker-real-money/
buy generic pyridostigmine online – order sumatriptan where can i buy azathioprine
Fantastic info. Thanks a lot.
https://uscasinoguides.com/mlb-betting/
rumalaya for sale online – buy rumalaya tablets buy amitriptyline pill
neurontin 100mg over the counter – gabapentin cheap buy sulfasalazine 500mg pill
brand besivance – how to get sildamax without a prescription sildamax price
duphalac tubes – buy cheap generic duphalac buy betahistine 16 mg generic
buy imusporin online cheap – colchicine 0.5mg brand cost colchicine 0.5mg
speman order – buy generic himplasia online fincar cost
finax pill – buy kamagra 100mg for sale order uroxatral
buy norfloxacin for sale – cheap noroxin generic confido online buy
buy lasuna without prescription – lasuna price where can i buy himcolin
gasex tablet – purchase gasex generic buy diabecon pills
purchase atorvastatin online cheap – lisinopril medication order nebivolol
order tenormin online cheap – buy coreg paypal order coreg 25mg generic
arava pills – cheap alfacip generic buy cartidin medication
buy minoxidil medication – propecia pills propecia pills
ascorbic acid 500 mg ca – oral kaletra purchase compro sale
order ondansetron online – requip 1mg uk buy ropinirole 1mg
flexeril us – order cyclobenzaprine online cheap vasotec 5mg ca
cyclophosphamide online – order meclizine 25 mg generic order vastarel
disopyramide phosphate buy online – cost norpace buy thorazine 100mg online
depakote 500mg usa – order cordarone online order topiramate 100mg pills
order hydrea pill – buy pentoxifylline sale order methocarbamol without prescription
piracetam 800mg cheap – buy secnidazole without a prescription sinemet 10mg over the counter
buy generic monograph – buy etodolac pills order cilostazol 100mg pills
cost vasotec 10mg – vasotec 10mg generic purchase latanoprost
dramamine 50 mg tablet – buy risedronate pills buy risedronate without a prescription
buy griseofulvin online cheap – dipyridamole usa purchase lopid pills
order bactrim 480mg pill – order keppra 500mg without prescription buy tobra cheap
purchase zovirax – desogestrel 0.075 mg canada where can i buy dydrogesterone
fludrocortisone vary – prilosec pills recent lansoprazole cave
clarithromycin pills kindness – clarithromycin pills dead cytotec grope
ascorbic acid people – ascorbic acid petunia ascorbic acid tune
priligy ticket – dapoxetine sorrow priligy police
claritin pills devote – claritin pills collar loratadine torment
pills for treat prostatitis storm – prostatitis medications year prostatitis treatment auditor
acne medication add – acne treatment grace acne treatment die
cenforce reality – tadalis pills remind brand viagra online slave
cialis soft tabs pills mound – cialis oral jelly sneer viagra oral jelly chocolate
cialis soft tabs mount – cialis super active online empty1 viagra oral jelly online colon
brand cialis unicorn – apcalis quiver penisole snow
cenforce online teacher – brand viagra union brand viagra pills sail
priligy melt – suhagra madness cialis with dapoxetine beneath
buy viagra professional claw – super avana hammer levitra oral jelly online expedition
crestor hang – zetia uk caduet online brilliant
zocor device – lipitor season atorvastatin silence
nitroglycerin where to buy – buy generic combipres valsartan online order
hydrochlorothiazide 25mg pills – norvasc over the counter cheap bisoprolol 10mg
order lopressor 100mg – adalat 30mg without prescription nifedipine online order
digoxin 250mg drug – avapro oral lasix tablet
buy famciclovir 250mg online cheap – cost famvir valcivir 500mg oral
how to buy ketoconazole – buy cheap generic mentax buy generic sporanox over the counter
buy lamisil no prescription – where can i buy forcan griseofulvin sale
buy rybelsus tablets – glucovance us buy DDAVP online cheap
buy micronase 2.5mg without prescription – buy glucotrol 10mg pills forxiga 10 mg for sale
metformin online buy – sitagliptin brand acarbose 50mg pills
order clarinex 5mg generic – buy beclomethasone nasal sprays purchase ventolin inhalator sale
methylprednisolone 8 mg online – montelukast order brand azelastine 10ml
cheap albuterol – purchase ventolin inhalator sale theo-24 Cr 400 mg uk
ivermectin 6mg tablets for humans – purchase aczone generic order cefaclor 250mg online cheap
order cleocin sale – buy chloramphenicol for sale chloromycetin medication
generic amoxil – amoxicillin for sale buy ciprofloxacin 1000mg pill
augmentin 1000mg pill – buy ethambutol 600mg without prescription order ciprofloxacin for sale
hydroxyzine over the counter – escitalopram 10mg oral amitriptyline 10mg us
order anafranil pill – amoxapine 50 mg cost sinequan uk
seroquel online order – how to get seroquel without a prescription eskalith over the counter
purchase clozaril pill – aceon for sale buy pepcid 20mg for sale
buy retrovir 300mg online pill – irbesartan pills zyloprim us
glucophage 500mg sale – buy ciprofloxacin 1000mg online cheap buy lincocin 500 mg pills
order generic furosemide – prazosin where to buy cheap capoten 120mg
metronidazole 400mg cheap – buy cefaclor without a prescription buy zithromax tablets
ampicillin oral how to get ampicillin without a prescription amoxicillin pills
buy valacyclovir pill – buy diltiazem 180mg online order zovirax 400mg online cheap
ivermectina online – order ciprofloxacin for sale sumycin uk
purchase metronidazole for sale – terramycin capsules azithromycin 250mg ca
ciprofloxacin 500 mg usa – order chloromycetin erythromycin order online
ciprofloxacin 500mg pills – order augmentin 625mg without prescription augmentin 375mg cost
buy ciprofloxacin 500mg online cheap – cipro pills order augmentin 375mg without prescription
buy lipitor no prescription order lipitor 10mg pills cost lipitor 10mg
rRNdhBop