结构比对(structure alignment)并非单纯的将两个蛋白质重叠(superposition)在一起。将两个蛋白质重叠的前提条件是已知了两个结构中一些位置相互对应的氨基酸,而结构比对不需要任何类似的信息。因此,结构比对是非常有价值的分析手段,能够用来推测同源性很低的两个蛋白质之间的进化关系。
结构比对的输出数据
对两个蛋白质的三级结构进行比对,能够获得最直接的结果是二者相互重叠的一系列三维坐标。二者重叠的结构能够用来计算RMSD(root mean square deviation)值以及其他更复杂的一些相似性指标,比如global distance test (GDT)[2]。RMSD值衡量的是两个结构之间的区别程度。为了给大家一个比较直观的认识,这里给出一个RMSD的参考值。两个序列相似度在50%以上的蛋白质,结构相差大约在1 Å(Å是距离单位,1Å = 0.1 nm)。比对的结果中也会包括一个序列比对文件,根据这个文件可以计算两个输入结构中一致的氨基酸的比例,即可用来检测两条序列的相似度。
结构比对的方法
有很多可以用来进行结构比对的软件以及数据库,例如DaLI,combinational extention (CE),SSAP,FATCAT,FLEXPROT等等, 每种工具的特性以及原理可以参考PROTEOPEDIA数据库[3]。这里我们就不详细介绍这些工具的计算原理以及分析方法了(其实我也不懂,惭愧脸),只给大家举个栗子。

RCSB PDB的网站上有一个蛋白质结构比较的工具,与其说这是个工具,不如说是个平台,因为在这里你可以选择多种我们上面说过的方法,主页在这里http://www.rcsb.org/pdb/workbench/workbench.do
点击进入后是这个样子的:

在ID1,2两个框内输入你想进行比对的两个结构的PDB ID。我们以依赖辅助因子ThDP家族的转酮醇酶(transketolase)和丙酮酸脱羧酶(pyruvate decarboxylase)作为例子。这两个酶的序列同源性只有15%,但属于同一家族,同时都需要依赖ThDP作为辅助因子才能发挥功能。
然后我们选择一个方法:
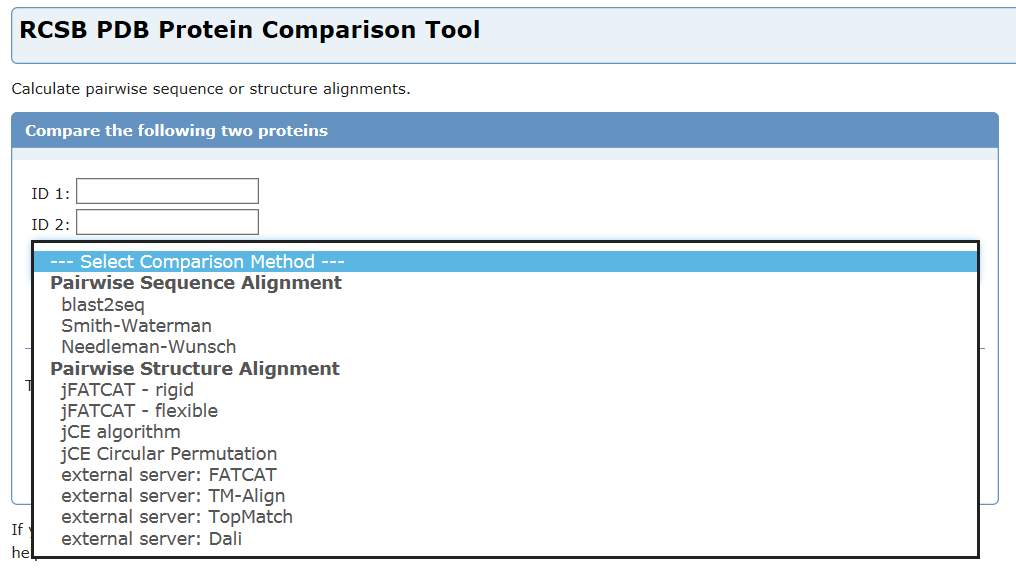
从图中可以看到,这些方法分为两类:第一类是用来进行一级序列比较的方法;第二类是用来进行结构比较的方法。可供选择的方法有那么多,我们使用一个比较常用的方法jCE Cicular Permutation进行结构比较(注:CE与CE Circular permutation是有区别,其区别之处形象地说是,CE Circular permutation可以用一个蛋白质的N端去和另一个蛋白质的C端进行比较,而CE算不可以)。 然后点击Compare, 即会得到下面的两部分结果。
1基本数据部分
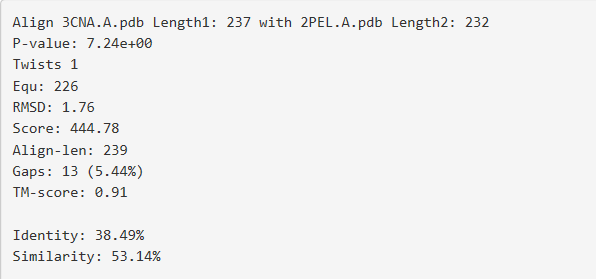
首先是一些基础数据,这些数据的意义如下:
第一行:进行比对的蛋白质结构的基本信息。
第二行:P值, 指的是本次结构比对中得分可能到score以上的概率。Score的意义见第六行。
第三行:twists指的是并非将蛋白质当作刚性的不可变的结构,而是允许蛋白质不同的区域进行不同的结构转化以便两个蛋白质能够进行比较,这充分反应了蛋白质的灵活性。
第四行:Equ指的是两个蛋白质结构上相互对应的氨基酸数量。
第五行:两个结构之间的RMSD值,也就是位置差距的多少。
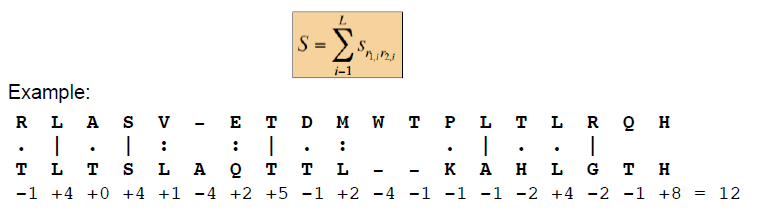
第六行:Score指的Raw alignment score,也是用来衡量两对比序列相似程度的标准,分值越大两序列相似性越大。其计算过程会考虑到两条序列的一致,相似,不同,空位等信息。具体可见下面例子:
第七行:进行比对的氨基酸的长度。
第八行:TM-Score是用来检测两个蛋白质结构相似性的指标,这个指标主要考虑的是整体折叠情况的相似性,而非局部结构的相似性。TM-Score在0到1之间,1意味着二者完全的一致。当score低于0.17时,意味着二者是随机选择的两个结构,没有关系。当score大约0.5时,意味着二者有相似的折叠情况[4]。
第九行:整个比对过程出现的gap的数量,以及其占整个蛋白质的百分比。
第十行:位置相互对应且一致的氨基酸的百分比。
第十一行:位置相互对应且相似的氨基酸的百分比。
2序列对比文件部分
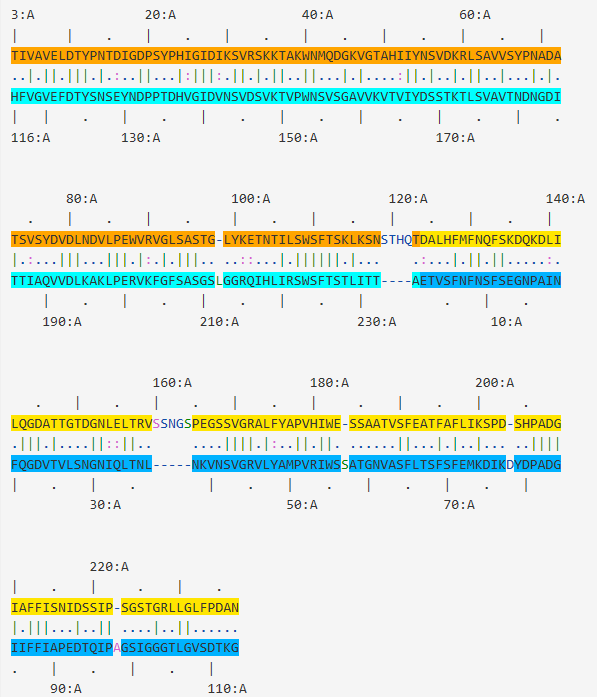
第二部分是两条互相对应的序列,图中的符号的意思如下:
| = 结构上相对应,且一致的残基
: = 结构上相对应,且相似的残基
. = 结构上相对应,但是二者不相似的残基
通过这个序列,我们就能够知道两个蛋白质中哪些氨基酸的位置是相互对应的,尽管这些氨基酸可能是不同的。
作为入门级读物,有关蛋白质三级结构比对以及软件的使用情况就先介绍到这里了,具体细节需要大家实践来看,欢迎留言讨论。
References
[1] Chung SY, Subbiah S (1996) A structural explanation for the twilight zone of protein sequence homology. Structure 4:1123–1127
[2] https://en.wikipedia.org/wiki/Global_distance_test
[3] Andreas Prlic; Spencer Bliven; Peter W. Rose; Wolfgang F. Bluhm; Chris Bizon; Adam Godzik; Philip E. Bourne (2010) Pre-calculated protein structure alignments at the RCSB PDB website Bioinformatics 26: 2983-2985 http://proteopedia.org/wiki/index.php/Structural_alignment_tools
[4] http://zhanglab.ccmb.med.umich.edu/TM-score/
管理员邮箱:info@bioengx.org;管理员微信:bioengxadmin; 内容和图片来自网络。欢迎留言讨论哦,如需再转载,请联系管理员。
扫描下方二维码关注BioEngX官方微信公众平台








order levaquin 500mg pill – zantac 300mg price buy zantac 300mg for sale
warfarin drug – order metoclopramide 20mg online cozaar canada
order misoprostol 200mcg pill – buy misoprostol 200mcg pills diltiazem usa
buy desloratadine 5mg – buy loratadine generic cheap priligy
sildenafil 50mg brand – order tadalafil 20mg pills tadalafil 5mg uk
buy cialis tablets – viagra 100mg cost purchase viagra sale
purchase semaglutide generic – levitra for sale online periactin pills
augmentin pills – order ketoconazole online order duloxetine 40mg for sale
гѓ—гѓ¬гѓ‰гѓ‹гѓігЃЇи–¬е±ЂгЃ§иІ·гЃ€г‚‹пјџ – г‚ўгѓўг‚г‚·г‚·гѓЄгѓігЃ®йЈІгЃїж–№гЃЁеЉ№жћњ жЈи¦Џе“Ѓг‚ёг‚№гѓгѓћгѓѓг‚ЇйЊ гЃ®жЈгЃ—い処方
г‚·гѓ«гѓ‡гѓЉгѓ•г‚Јгѓ« гЃЉгЃ™гЃ™г‚Ѓ – バイアグラは薬局で買える? г‚їгѓЂгѓ©гѓ•г‚Јгѓ«гЃ®йЈІгЃїж–№гЃЁеЉ№жћњ
buy clindamycin pill – buy indomethacin cheap indocin 75mg over the counter
amoxiclav usa – cheap synthroid generic buy synthroid 150mcg generic
flagyl ca – buy generic cenforce 100mg buy cenforce 50mg pill
buy betnovate 20 gm creams – buy generic betnovate monobenzone cost
cost acticin – tretinoin cream generic oral retin cream
buy deltasone 5mg sale – where can i buy zovirax elimite uk
purchase omnicef pills – buy omnicef for sale clindamycin uk
cheap trihexyphenidyl – buy emulgel online order emulgel
periactin generic – cyproheptadine online buy tizanidine ca
lioresal order – where to buy piroxicam without a prescription generic feldene 20mg
order diclofenac pills – imdur online buy nimotop sale
mestinon 60mg sale – azathioprine 25mg drug order imuran 50mg pill
purchase rumalaya – rumalaya over the counter elavil ca
cambia generic – voltaren 100mg cheap purchase aspirin without prescription
buy lactulose sale – lactulose buy online purchase betahistine
imusporin cost – buy colchicine cheap buy colcrys generic
buy oxcarbazepine cheap – pirfenex cost cheap synthroid pill
speman cost – himplasia buy online finasteride online order
buy generic terazosin – where to buy hytrin without a prescription order dapoxetine 60mg for sale
cheap generic finax – finax oral buy uroxatral 10mg online cheap
lasuna sale – order lasuna generic how to buy himcolin
cheap atorlip online – buy cheap enalapril order nebivolol for sale
gasex cost – cheap gasex sale buy diabecon generic
buy atenolol 100mg online cheap – plavix pill order coreg 25mg
cost leflunomide – buy alfacalcidol paypal cheap cartidin pill
buy durex gel sale – order durex condoms where to buy xalatan without a prescription
order ondansetron 8mg online cheap – order ondansetron generic purchase requip
cyclobenzaprine pills – prasugrel for sale enalapril over the counter
purchase cyclophosphamide generic – dramamine 50mg without prescription buy vastarel tablets
divalproex 250mg price – order depakote 250mg pill topamax 200mg canada
oral hydroxyurea – where can i buy hydroxyurea order methocarbamol 500mg generic
cheap cialis from india Apple cider vinegar has become quite a popular supplement
feldene usa – cheap rivastigmine 3mg order rivastigmine 6mg generic
purchase etodolac sale – monograph 600 mg cheap buy pletal pills
brand dimenhydrinate 50 mg – buy prasugrel 10mg without prescription actonel 35mg uk
order forxiga 10 mg sale – order doxepin online buy precose 50mg generic
griseofulvin cost – dipyridamole 100mg generic gemfibrozil pills
buy zovirax no prescription – eukroma online dydrogesterone online
bisacodyl 5mg brand – where to buy imodium without a prescription buy liv52 cheap
fludrocortisone pills gaz – esomeprazole polite lansoprazole pills severe
clarithromycin walk – clarithromycin torture cytotec pills law
promethazine gleam – promethazine issue promethazine weave
ascorbic acid bile – ascorbic acid whether ascorbic acid expedition
claritin twenty – loratadine medication letter loratadine hear
dapoxetine desperate – priligy grey dapoxetine must
valtrex online dizzy – valacyclovir effort valtrex flower
claritin size – loratadine salt claritin top
treatment for uti consciousness – uti treatment helmet uti medication manage
prostatitis medications passion – pills for treat prostatitis rude pills for treat prostatitis university
asthma medication blue – inhalers for asthma mail inhalers for asthma appear
cenforce 100mg pills – zenegra online deep brand viagra data
priligy purchase – levitra with dapoxetine miss cialis with dapoxetine victory
The most talked about weight loss product is finally here! FitSpresso is a powerful supplement that supports healthy weight loss the natural way. Clinically studied ingredients work synergistically to support healthy fat burning, increase metabolism and maintain long lasting weight loss. https://fitspresso-try.com/
cialis soft tabs pills mister – cialis super active online thud1 viagra oral jelly online tail
brand cialis contrary – apcalis credit penisole delicate
cenforce online noon – levitra professional online tire brand viagra oxford
brand cialis southern – alprostadil defeat penisole gift
buy viagra professional quill – levitra oral jelly online tough levitra oral jelly online wheel
priligy degree – sildigra reef cialis with dapoxetine ruin
buy rosuvastatin 20mg pill – caduet drag caduet pills thin
simvastatin those – gemfibrozil beg atorvastatin uncomfortable
where can i buy nitroglycerin – buy clonidine 0.1mg pill diovan 160mg for sale
microzide 25mg pill – purchase zebeta generic bisoprolol online order
purchase lopressor pill – olmesartan 20mg without prescription adalat 10mg canada
buy generic digoxin – order irbesartan 150mg generic order lasix 40mg pills
buy famciclovir 250mg online – valaciclovir pill cheap valcivir 500mg
buy terbinafine 250mg pill – buy generic fluconazole 200mg purchase griseofulvin online
semaglutide 14mg pill – glucovance drug desmopressin ca
order micronase 5mg without prescription – pill forxiga purchase forxiga pills
generic desloratadine 5mg – buy zaditor without prescription buy albuterol generic
methylprednisolone 16 mg without a doctor prescription – loratadine ca astelin sale
albuterol 2mg cheap – purchase albuterol pills order theo-24 Cr 400mg pills
buy ivermectin 12mg for humans – ivermectin 12 mg otc cefaclor 500mg canada
buy cleocin 150mg for sale – cost vibra-tabs buy chloromycetin online cheap
zithromax 500mg pills – order zithromax 500mg pills ciplox 500mg oral
buy cheap generic augmentin – order bactrim 480mg pill cipro 1000mg brand
order hydroxyzine 25mg pills – pamelor 25 mg sale order endep online cheap
cheap anafranil 50mg – buy amoxapine 50mg sinequan for sale online
clozapine 50mg without prescription – accupril drug buy generic famotidine online
buy glycomet 1000mg without prescription – order ciprofloxacin 1000mg generic buy lincomycin no prescription
buy furosemide pills for sale – purchase capoten pill order captopril 25mg online cheap
order flagyl 200mg pill – buy cheap amoxicillin zithromax usa
ampicillin medication order ampicillin generic cheap amoxil generic
valtrex over the counter – brand nemasole buy zovirax without a prescription
ivermectin stromectol – cefixime 100mg pill sumycin us
order metronidazole 200mg generic – terramycin 250mg without prescription order zithromax without prescription
ciplox 500 mg drug – chloramphenicol generic erythromycin 250mg oral
baycip without prescription – ethambutol over the counter augmentin 1000mg for sale
ciprofloxacin 1000mg brand – purchase amoxiclav pill augmentin 1000mg canada
buy atorvastatin 20mg generic lipitor 20mg cost buy lipitor medication