已知蛋白质的一级序列,如何能够获得其三级结构呢?结构生物学领域的科学家们能够利用扫描电镜,核磁共振以及X衍射等手段获得绝大多数蛋白质的结构。但解析蛋白质结构毕竟是一个专门的领域,对于“门外汉”,同时又没有资金委托他人来解析结构的研究者来说,就只能自己预测了。
预测分为从头预测和同源建模。同源建模预测的准确度高于从头预测,尽管依然没有真实测定来的准确。通过对预测结构的分析, 是可以获得很多信息的,比如解释酶的催化机理或者预测蛋白质的突变位点, 推断蛋白质结构与功能的关系等等。那么如何利用同源建模来预测蛋白质的三级结构呢?
同源建模的原理
首先我们要了解同源建模的原理。同源建模具体指的是以与未知结构蛋白具有同源性的已知结构的蛋白作为模版,用生物信息学的方法通过计算机模拟和计算,根据一级序列预测其三维空间结构。
同源建模基于两个原理。第一,一个蛋白质的结构由其氨基酸序列唯一决定,知道其一级序列,在理论上就可以获取其二级结构以及三级结构。第二,蛋白质的三级结构在进化中更稳定或者说更保守。如果两个蛋白质的氨基酸序列有50%相同,那么约有90%的a-碳原子的位置偏差不超过3 Å,这是同源模型化方法在结构预测方面成功的保证。同源建模通常要求模板蛋白与目标蛋白的序列一致性高于30%。
同源建模的步骤
同源建模可以通过以下7个步骤进行[1]:
1、搜索结构模型的模板(T)。
同源建模方法假设两个同源的蛋白质具有相同的骨架。为待预测的蛋白质建立模型时,首先按照同源蛋白质的结构建立模板T。所谓模板是一个已知结构的蛋白质,该蛋白质与目标蛋白质U的序列非常相似。如果找不到这样的模板,则无法运用同源模型法。
2、序列比对。
将目标蛋白质U的序列与模板蛋白质T的序列进行比对,使U的氨基酸残基与模板蛋白质的残基匹配。比对中允许插入和删除操作。
3、建立骨架。
将模板结构的坐标拷贝到目标U,仅拷贝匹配残基的坐标。在一般情况下,通过这一步建立目标蛋白质U的骨架。
4、构建目标蛋白质的侧链。
可以将模板相同残基的坐标直接作为目标蛋白质的残基坐标,但是对于不完全匹配的残基,其侧链构象是不同的,需要进一步预测。侧链坐标的预测通常采用已知结构的经验数据,如ROTAMERS 数据库中的经验结构数据。
5、构建目标蛋白质的环区(loop)。
在第2步的序列比对中,可能加入空位,这些区域常常对应于二级结构元素之间的loop,对于环区需要另外建立模型。一般也是采用经验性方法,从已知结构的蛋白质中寻找一个最优的环区,拷贝其结构数据。如果找不到相应的环区,则需要用其它方法。
6、优化模型。
通过上述过程为目标蛋白质U建立了一个初步的结构模型,在这个模型中可能存在一些不相容的空间坐标,因此需要进行改进和优化,如利用分子动力学、模拟退火等方法进行结构优化。
7、模型验证
蛋白质结构预测结果需要进行验证。下面是一个常用的结构检测服务器:来源于UCLA-DOE的SAVES服务器,http://services.mbi.ucla.edu/SAVES/
这个服务器内包括6个用于预测结构准确性的工具,分别是PROCHECK、WHAT_CHECK、ERRAT、VERIFY_3D、PROVE、CRYST1 record matches、Ramachandran Plot、WedMol Viewer。这些软件可以分为两类:一类是检查结构的立体化学信息,比如PROCHECK。立体化学信息包括键长,键角,对称性,结构包埋率等等。另一类是检查序列与结构的匹配度,比如VERIFY_3D。用户可以同时运行所有的软件,也可以单独选择某一个进行运行。
下面举一个例子,说明软件PROCHECK预测结构准确性的原理。
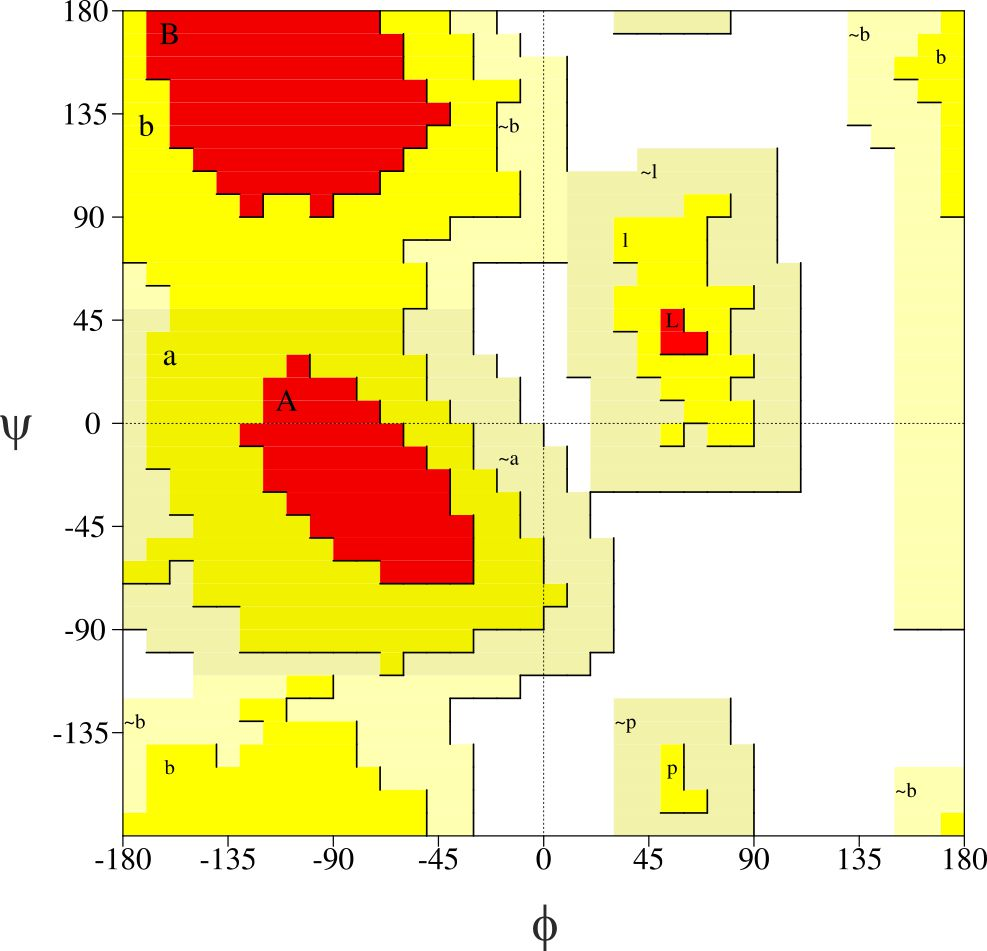
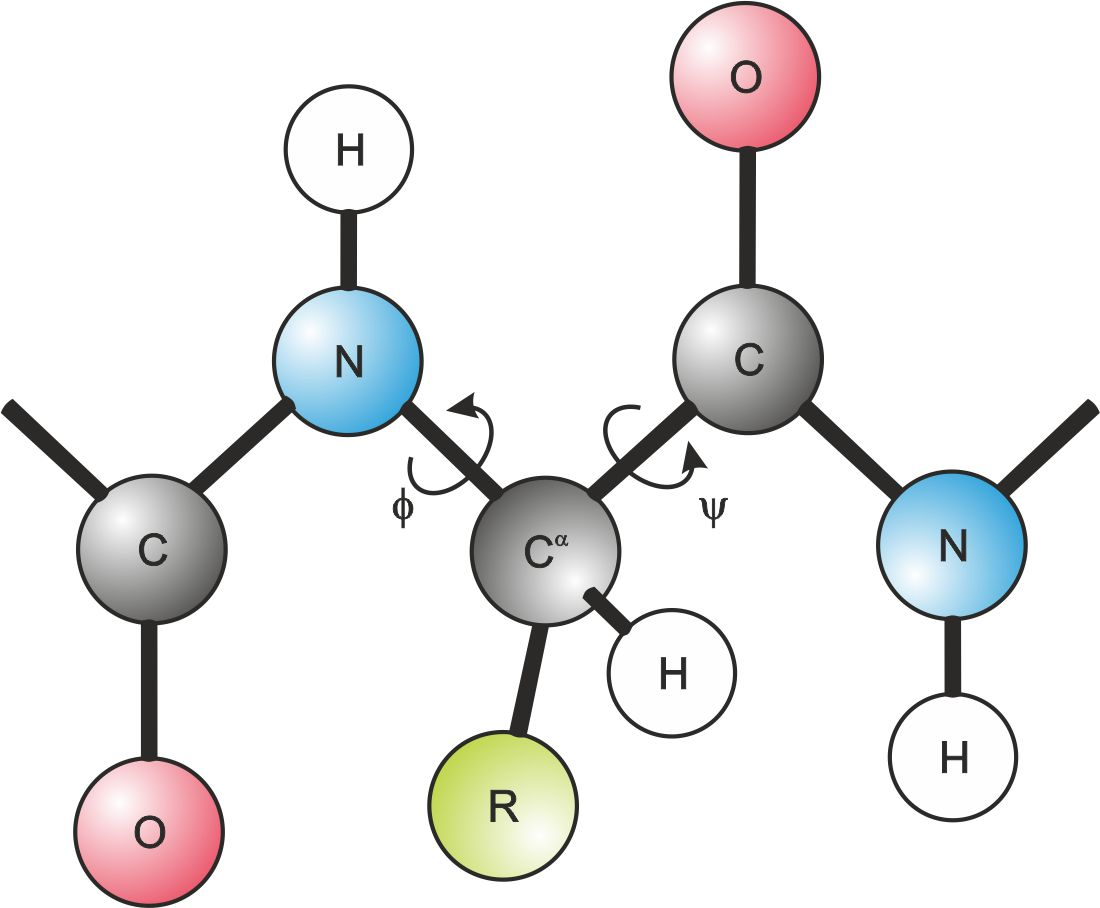
上图是PROCHECK生成的拉氏图(Ramachandran plot)。拉氏图就是蛋白质中残基的两个二面角ϕ 和 ψ 的相互关系图,将所有氨基酸的ϕ 和 ψ值相互对应,就可以做出这样的图了。在一个氨基酸中,ϕ指的是Ca与N之间的二面角, ψ指的是Cα与羧基C之间的二面角(详见下图)。
拉氏图分为四个区域: most favoured (red), additional allowed (yellow), generously allowed (light yellow) 以及disallowed (white),红色表示的是最合适的区域,白色表示最不适合的区域。一个高质量的蛋白质结构的分辨率通常小于2 Å, R-factor小于20%。统计分析118个这样的蛋白质结构表明,对于一个高质量的模型结构来说,其90%的ϕ and ψ 值都应该在红色的区域内[2]。
同源建模的工具
有很多软件或服务器能够基于目标蛋白的序列进行同源建模。基于可靠性,软件运行速度,以及结构相似性三个方面,很多研究对这些软件进行了比较,具体可参考文献3-5。
下面对几个常用的软件进行简单介绍:
Swiss-Model
这是非专业人士使用最广泛的一个工具,其特点是简单,自动化并且对学术团队免费。
网址:https://swissmodel.expasy.org
Modeller
该软件由Sali lab开发,是非常常用的一个同源建模工具,目前最新版本是9.19,可在window和linux操作系统下运行。可以进行多聚体建模,二硫键建模,杂原子建模等。自带一套结构评价体系。该工具完全通过命令行控制,操作略复杂。
网址:https://salilab.org/modeller/
zhang服务器
Zhang服务器是使用I-TASSER算法用于蛋白质结构和功能预测的在线平台。
网址:https://zhanglab.ccmb.med.umich.edu/I-TASSER/
Yasara
一款商业化的多功能软件,内部包括同源建模模块。
网址:http://www.yasara.org/homologymodeling.htm
nest
nest由美国Columbia University 的Barry Honig教授及其同事开发,能够在SGI and Intel Linux平台下运行。
网址:http://honig.c2b2.columbia.edu/nest/
参考文献
[1]《生物信息学基础》第七章,作者:孙啸,陆租宏,谢建明。清华大学出版社
[2] Laskowski R A, MacArthur M W, Moss D S, et al. PROCHECK: a program to check the stereochemical quality of protein structures[J]. Journal of applied crystallography, 1993, 26(2): 283-291.
[3] Nayeem A, Sitkoff D, Krystek S Jr. A comparative study of available software for high-accuracy homology modeling: From sequence alignments to structural models. Protein Sci. 2006;15:808–24.[PMC free article] [PubMed]
[4] Wallner B, Elofsson A. All are not equal: A benchmark of different homology modeling programs. Protein Sci. 2005;14:1315–27
[5] Vyas V K, Ukawala R D, Ghate M, et al. Homology modeling a fast tool for drug discovery: current perspectives[J]. Indian journal of pharmaceutical sciences, 2012, 74(1): 1.