酶的结构决定功能,这通常是酶工程中的关键思想,其中的目标就是理解结构和功能关系,而最主要的就是获得酶的结构。当前Pfam中有14,849个蛋白家族(1)在具有50个或更多个残基的数据库中,其中4752个家族至少有一个成员具有实验确定的X射线晶体或核磁共振(NMR)结构,还有额外的3984个家族,可以建立可靠的比较模型。也就是说共有8736个蛋白家族可以通过同源建模的方式获得可靠的蛋白结构。
当前建模的方法有很多种,常用的如Swiss-model,Modeller,YASARA,I-TASSER等。在本研究中,CPCR2作为我们的研究对象(UniProtKB数据库登录号O42703),CPCR2是来自近平滑假丝酵母的羰基还原酶(Carbonyl Reductase from Candida parapsilosis)。我们的目标是用Modeller,YASARA,I-TASSER三种比较公认建模可靠的方法构建,验证和比较CPCR2的同源性模型。
同源性建模的基本要求是与蛋白质数据库(PDB)中可用的已知3D晶体结构的序列一致度大于30%。同源建模中涉及的步骤是:
1)模板选择
2)靶-模板比对(BLASTP,ClustalW)
3)三维结构建模
4)模型验证(Ramachandran plot,Errat plot,Prosa energy plot,Verify 3D plot,Z-score,3D packing,dihedral angles)
5)活性位点表征(分子对接)
使用Modeller9.11构建同源模型
从UniProtKB数据库(登录号O42703)检索CPCR2的氨基酸序列。使用针对蛋白质数据库(PDB)的基本局部搜索比对工具(BLAST)进行序列相似性搜索。结果,获得了25个命中,但是与CPCR2的序列一致度低于35%。因此,当前的情况同源建模是相当具有挑战性的。在这种情况下,优选的方式是去寻找多个模板,但是模型的可靠性成为主要问题。在25个命中中,来自Sulfolobus tokodaii菌株7(PDB ID:2EER)的NAD-依赖的醇脱氢酶与CPCR2具有35%的序列一致度,然而,在活性位点中不存在催化性锌,并且查询覆盖率也低。来自酵母醇脱氢酶I,酿酒酵母发酵酶(PDB ID:2HCY)的另一个PDB,其具有31%的序列一致度和99%的查询覆盖度,其满足所有标准。然而,在BLAST搜索过程中,另一种晶体结构出现了,绿脓杆菌醇脱氢酶(PDB ID:1LLU)的三元复合物,其也满足与2HCY相同的标准。因此,最后我们选择1LLU作为使用Modeller9.11的同源性建模的起始模板。来自1LLU的CPCR2序列和模板的比对使用CLUSTALW2在线服务器进行。CPCR2与来自铜绿假单胞菌(Pseudomonas aeruginosa)的同源蛋白ADH(PDB ID:1LLU)的序列比对具有30%的序列一致度,如图1所示:

Figure 1 Sequence alignment of target CPCR2 and template sequence (PDB ID: 1LLU). ESPript3 outputs obtained with the sequences from the SWISSPROT databank and alignment with CLUSTALW2.
最初通过在Modeller9.11中使用自动模型建立选项产生100个CPCR2的粗同源模型。选择具有最低DOPE评分的20个模型和molpdf评分进行进一步的细化。然后使用Ramachandran图,在每个细化步骤的ERRAT图检查所选择的模型的二面角分布。使用Modeller9.11 loop建模进行loop区域细化。在ERRAT图中未显示良好结果的残基提交进行能量最小化。优化的模型进行分析并进行进一步优化,直到获得令人满意且相当精确的结构。最终优化好的模型如图2所示。
模建的酶是同源二聚体复合物,其中含有催化和结构锌。 它还显示NAD(H)结合结构域为Rossman折叠,和底物结合口袋连在一起。 模板和目标模型之间的RMSD为0.193Å。此外,使用Ramachandran图评估构建的3D模型的立体化学。 模型的Ramachandran图显示86.40%的残留在最有利的区域,11.7%在允许的区域,而0.5%的残留在不允许的区域,强烈表明当前的模型质量比较好。ERRAT检查非键合原子相互作用的总体质量因子,值越高,质量越高。 对于良好模型的质量,ERRAT图的普遍接受值大于50。 ERRAT服务器预测的总体质量因子为84.38。 来自VERIFY_3D服务器的结果显示,CPCR2的95.25%的残基具有> 0.2的平均3D-1D得分,从而验证构建的同源性模型的质量良好。
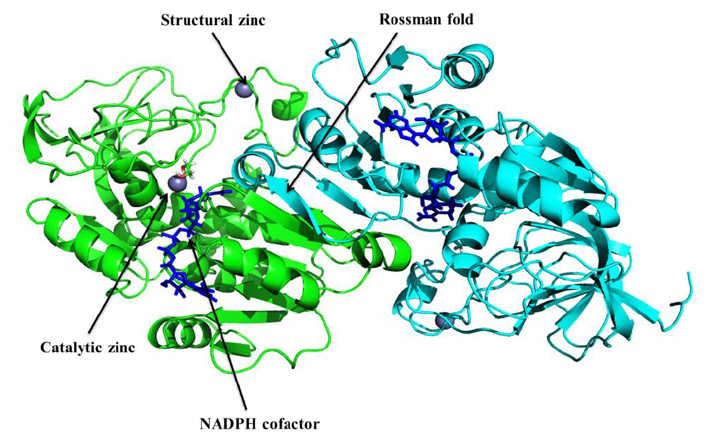
Figure 2 Homology model of CPCR2 constructed by using Modeller9.11. It is a dimeric structure consisting of two monomeric units, zinc ions (catalytic and structural zinc) are shown as spheres and cofactor is in blue sticks.
使用Yasara构建同源模型
使用Yasara用5种不同模板(PDB ID:1LLU,PDB ID:2EEZ,PDB ID:3JV7,PDB ID:2HCY,PDB ID:1H2B)构建杂交同源模型。构建的杂交模型是基于profile-profile比对的二聚体:在与模板1LLU的最佳比对中,将336个目标残基(92.0%)的CPCR2中的309个与模板残基比对。 在这些比对残基中,序列同一性为31.7%,序列相似性为47.6%。在杂交同源模型(图3所示)中,大多数氨基酸序列从PDB ID:1LLU(图3中以蓝色显示)复制。 该模型的总体质量令人满意,并且具有-1.171的Z-score打分。 Modeller模型和Yasara模型显示存在两个锌; 一种是催化的,另一种是结构Zn2+。
Table 2 Overall quality of hybrid homology model of CPCR2 constructed using Yasara
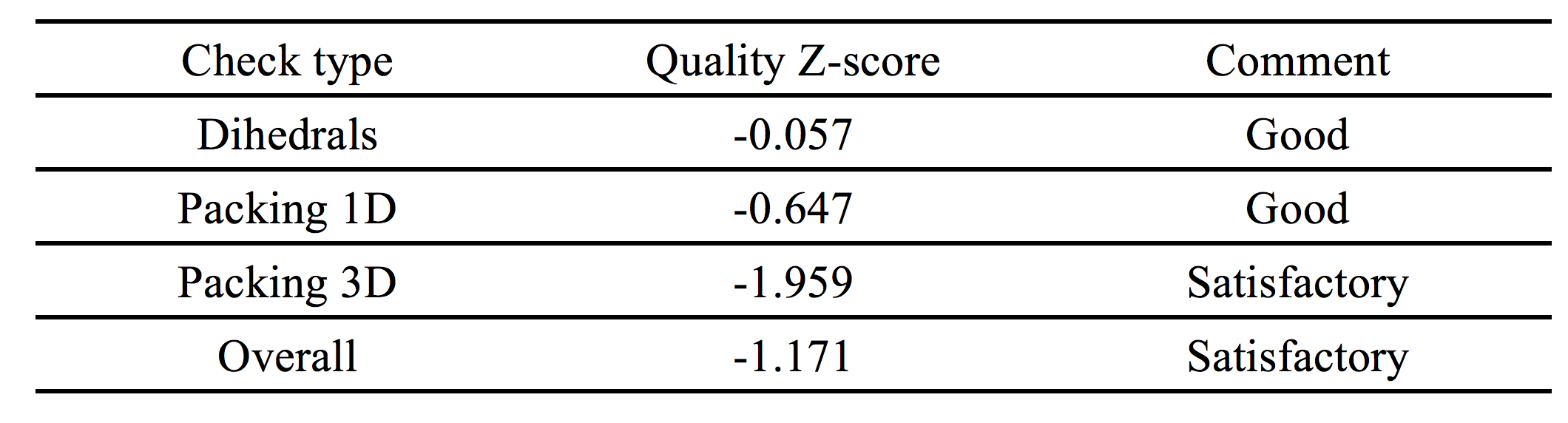
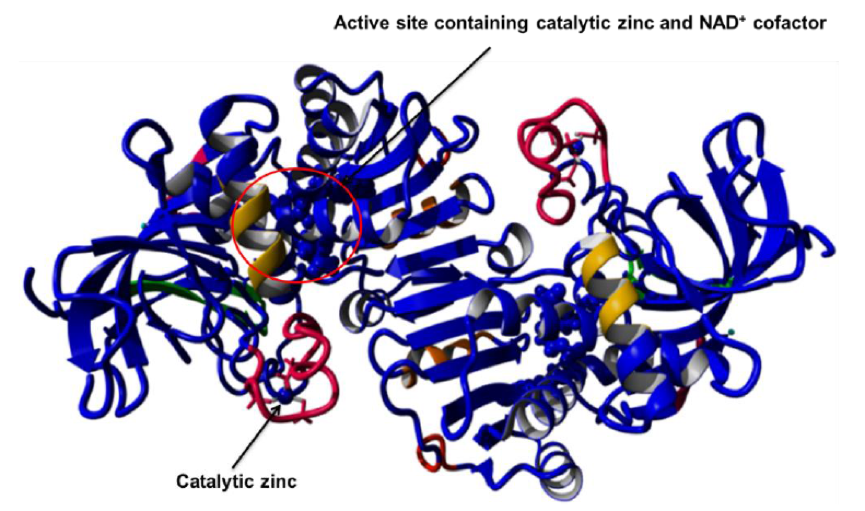
Figure 3 Hybrid homology model constructed using Yasara. Different colour represents different templates used in this study. Most of the part (blue colour) was taken from 1LLU template.
使用Zhang服务器构建同源模型
Zhang服务器是使用I-TASSER算法用于蛋白质结构和功能预测的在线平台。 它在CASP7和CAST8中排名第一。 将CPCR2的氨基酸序列提交给服务器(http://zhanglab.ccmb.med.umich.edu/I-TASSER/)。 获得Z-得分排名的前5个模型。 获得具有两个亚基的二聚体模型,其排列偏离的典型的MDR的Rossmann折叠(如图4所示)
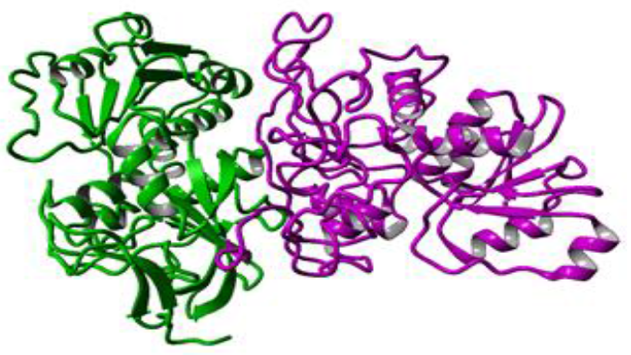
Figure 4 Homology model constructed using Zhang server. The constructed model is a dimer without anti-parallel Rossman folds.
单体A看起来相当好,但仍然有螺旋和β折叠的位移,而单体B完全变形。 与单体A相比,在单体B中存在非常少的α螺旋和β折叠。在单体B也没有Rossman折叠的特征。在两个单体亚基之间的片层在模型中不再存在(参见图4)。 因此,这个模型对进一步的研究没有用。
CPCR2同源性模型之间的比较
除了来自Zhang服务器,所有模建的同源性模型看起来相似并且再现典型的MDR酶的二聚体Rossman折叠,但是在它们的结构水平上具有一些特征性差异。
为了分析差异,两个同源模型在Yasara中使用Align模块进行结构比对,发现Cα原子的RMSD为1.294 Å,残基100%比对。而只比对A_chain,发现RMSD为1.292 Å,并且仅336个氨基酸中的313个比对。类似地,在只比对B_chain的情况下,发现RMSD为1.193 Å,并且在336个氨基酸中仅有307个对齐。发现模型的活性位点残基存在显著差异。通过使用Modeller产生的模型中的半胱氨酸残基处于标准质子化状态并且不与催化性锌离子配位。除此之外,模型包含与辅因子NADPH组合的醇底物,其在醇类底物的情况下应该存在的是氧化态的NAD +。在范围从57-68和231-257的氨基酸之间存在另一个显著差异(参见图5),其中活性位点残基His65在两个模型中不同,并且使用Modeller生成的模型中不与催化性锌离子结合。因此,我们决定进一步使用Yasara构建的混合模型。
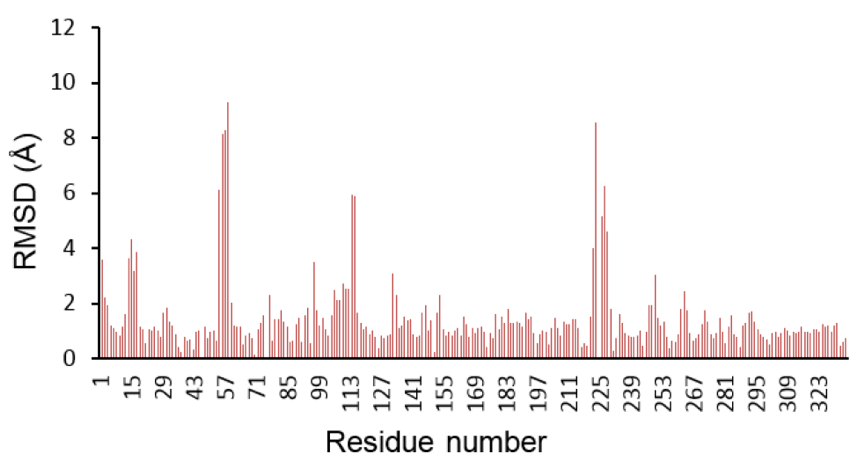
Figure 5 Difference between RMSD C-alpha carbon atoms per residue of Modeller model and Yasara hybrid model.
在本研究中,CPCR2(PDB ID:4C4O)的X射线晶体结构已经被解出。因此,我们比较了最终选择的杂交Yasara模型与CPCR2的实验晶体结构。所构建的杂交同源模型和CPCR2的晶体结构之间的总RMSD为1.96 Å,具有98.25%序列一致性。此外,发现催化锌离子的活性位点残基的RMSD为5 Å以内。为了比较,我们仅考虑晶体结构的单体B链,因为其在结晶过程中良好地解析。我们观察到存在结构锌的loop区域中的明显的差异。除此之外,锌的所有配位残基(Cys44,His65和Asp154)在两个结构中彼此良好对齐。
总结
总之,我们已经构建并验证了CPCR2的三维结构。 我们发现,Yasara杂交同源性建模与基于单一模板的建模方法,如Modeller和I-Tasser相比,给出更好的结果。 因此,我们可以得出结论,对于低序列同一性 < 40%的模板建模,YASARA的杂交同源性建模似乎是有希望的。






В этой публикации мы сосредоточимся на интересных аспектах одной из самых актуальных тем современности. Совмещая факты и мнения экспертов, мы создадим полное представление о предмете, которое будет полезно как новичкам, так и тем, кто глубоко изучает вопрос.
Получить дополнительные сведения – https://vyvod-iz-zapoya-1.ru/
Can you tell us more about this? I’d want to find out some additional information.
Thanks for the information you brought to us. It is very interesting and new. Looking forward to reading more useful and new articles from you.
kabargresik.com
You actually make it seem so easy with your presentation but I find this topic to be actually something that I believe I would never understand. It kind of feels too complex and very broad for me. I’m taking a look ahead on your next publish, I will attempt to get the hang of it!
[url=http://emeacc.org/xbet-les-paris-sportifs-les-plus-populaires-en-ligne/]emeacc.org[/url]
В этой медицинской статье мы погрузимся в актуальные вопросы здравоохранения и лечения заболеваний. Читатели узнают о современных подходах, методах диагностики и новых открытий в научных исследованиях. Наша цель — донести важную информацию и повысить уровень осведомленности о здоровье.
Подробнее – https://vyvod-iz-zapoya-17.ru/
The helmet of the man’s cock weaves a picture of no recognition as the father’s cum streaks itself through the watery haze.
“It was intense. Really intense. We ran so many drills. I am exhausted.” He explains.
“I don’t wanna jack-off, dad.” He says flabbergasted to his dad as he turns off the tap to the hot and chilly water in the shower. “That you are, son. You are busting at the seams with your youth and muscles. Rippled from those vigorous physical workouts and stroking sessions. I bet.” His dad says. “You are gonna hafta to take care of that or you are gonna be miserable. You know that son, doncha. You know, I am right.”
Daddy gave me a look implying his shoes are needed to be removed. “Can I take off your loafers, daddy ?” I asked so quietly. He looked at me and nodded. I took off both his loafers and put them away neatly in front of me. Daddy then raised his right leg and stepped on my forehead, pushing me down into his loafers. It is a mixed smell with sweat, testosterone, and leather. I couldn’t help but take a big whiff every time I breathed. “Hands-on the floor” he commanded. He moved his feet from my head to my hands. “Kiss them and make the stink goes away using only your fag tongue.” “Yes sir.” “Pick it up and smell the crotch.” His dad says.
His father kicks the shoes from his feet and slides his khaki pants off, while still holding, the resistant nut-sac of his virile son in his right hand. He is now as naked as his 17-year-old son. [url=https://arturzasada.pl/]dzieciД™ce porno[/url] His dad takes a seat upon the closed commode lid.
Valuable material. Regards!
https://uscasinoguides.com/north-carolina-casinos/
c3a05e